UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-K
(Mark One)
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2020
OR
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ___________ to ___________
Commission File Number: 001-39971
Landos Biopharma, Inc.
(Exact Name of Registrant as Specified in its Charter)
|
Delaware |
81-5085535 |
|
( State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer |
|
|
|
|
1800 Kraft Drive, Suite 216 Blacksburg, Virginia |
24060 |
|
(Address of principal executive offices) |
(Zip Code) |
Registrant’s telephone number, including area code: (540) 218-2232
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
Common stock, par value $0.01 per share |
|
LABP |
|
The Nasdaq Stock Market LLC |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☐ |
|
|
|
|
|
|||
|
Non-accelerated filer |
|
☒ |
|
Smaller reporting company |
|
☒ |
|
|
|
|
|
|
|
|
|
|
|
|
|
Emerging growth company |
|
☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The registrant was not a public company as of the last business day of its most recently completed second fiscal quarter and therefore cannot calculate the aggregate market value of its voting and non-voting common equity held by non-affiliates as of such date.
As of March 29, 2021, the registrant had 40,117,598 shares of common stock, $0.01 par value per share, outstanding.
|
|
|
Page |
|
|
|
|
|
Item 1. |
1 |
|
|
Item 1A. |
76 |
|
|
Item 1B. |
117 |
|
|
Item 2. |
117 |
|
|
Item 3. |
117 |
|
|
Item 4. |
117 |
|
|
|
|
|
|
Item 5. |
118 |
|
|
Item 6. |
118 |
|
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operation |
119 |
|
Item 7A. |
128 |
|
|
Item 8. |
128 |
|
|
Item 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
128 |
|
Item 9A. |
128 |
|
|
Item 9B. |
128 |
|
|
|
|
|
|
Item 10. |
129 |
|
|
Item 11. |
131 |
|
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
134 |
|
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
137 |
|
Item 14. |
139 |
|
|
|
|
|
|
Item 15. |
140 |
|
|
Item 16. |
140 |
|
|
141 |
||
SPECIAL CAUTIONARY NOTICE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K, or this Annual Report, contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, that involve substantial risks and uncertainties. The forward-looking statements are contained principally in Part I, Item 1. “Business,” Part I, Item 1A. “Risk Factors,” and Part II, Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” but are also contained elsewhere in this Annual Report. In some cases, you can identify forward-looking statements by the words “may,” “might,” “will,” “could,” “would,” “should,” “expect,” “intend,” “plan,” “objective,” “anticipate,” “believe,” “estimate,” “predict,” “project,” “potential,” “continue” and “ongoing,” or the negative of these terms, or other comparable terminology intended to identify statements about the future. These statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. Although we believe that we have a reasonable basis for each forward-looking statement contained in this Annual Report, we caution you that these statements are based on a combination of facts and factors currently known by us and our expectations of the future, about which we cannot be certain. Forward-looking statements include statements about:
|
|
• |
the timing, progress and results of our clinical trials of BT-11, NX-13 and any other product candidates, including statements regarding the timing of initiation and completion of studies or trials and related preparatory work, the period during which the results of the trials will become available and our research and development programs; |
|
|
• |
the timing of any submission of filings for regulatory approval of, and our ability to obtain and maintain regulatory approvals for, BT-11, NX-13 and any other product candidates for any indication; |
|
|
• |
our expectations regarding the size of the patient populations, market acceptance and opportunity for and clinical utility of our product candidates, if approved for commercial use; |
|
|
• |
our manufacturing capabilities and strategy, including the scalability and commercial viability of our manufacturing methods and processes; |
|
|
• |
our expectations regarding the scope of any approved indication for BT-11, NX-13 or any other product candidate; |
|
|
• |
our ability to successfully commercialize our product candidates; |
|
|
• |
our ability to leverage our LANCE platform to identify and develop future product candidates; |
|
|
• |
our estimates of our expenses, ongoing losses, future revenue, capital requirements and our needs for or ability to obtain additional funding; |
|
|
• |
our ability to establish or maintain collaborations or strategic relationships; |
|
|
• |
our ability to identify, recruit and retain key personnel; |
|
|
• |
our ability to protect and enforce our intellectual property position for our product candidates, and the scope of such protection; |
|
|
• |
our financial performance; |
|
|
• |
our competitive position and the development of and projections relating to our competitors or our industry; |
|
|
• |
the impact of laws and regulations; |
|
|
• |
the impact of the COVID-19 pandemic; and |
|
|
• |
our expectations regarding the time during which we will be an emerging growth company under the JOBS Act. |
You should refer to “Item 1A. Risk Factors” in this Annual Report for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all. The forward-looking statements in this Annual Report represent our views as of the date of this Annual Report. We anticipate that subsequent events and developments may cause our views to change. However, while we may elect to update these forward-looking statements at some point in the future, we undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law. You should, therefore, not rely on these forward-looking statements as representing our views as of any date subsequent to the date of this Annual Report.
You should read this report and the documents that we reference in this report, completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
All brand names or trademarks appearing in this Annual Report are the property of their respective owners. Solely for convenience, the trademarks and trade names in this Annual Report are referred to without the symbols ® and TM, but such references should not be construed as any indication that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto.
Unless the context requires otherwise, references in this report to “Landos,” the “Company,” “we,” “us,” and “our” refer to Landos Biopharma, Inc. and its subsidiaries.
We are a clinical-stage biopharmaceutical company, founded in 2017, focused on the discovery and development of oral therapeutics for patients with autoimmune diseases that are the first to target new mechanisms of action, including the LANCL2, NLRX1 and PLXDC2 immunometabolic pathways. Our core expertise is in the development of therapeutic candidates targeting novel pathways at the interface of immunity and metabolism. Based on our understanding of the role that a cell’s metabolic pathways have on modulating inflammatory responses, we aim to inhibit these inflammatory responses by changing the metabolic processes in target cells. We leverage our proprietary AI-based precision medicine platform, our LANCE platform, to identify novel therapeutic targets based on predictions of immunometabolic function and create therapeutic candidates to engage those targets in areas of unmet medical need. Through our LANCE platform, we have identified seven novel immunometabolic targets and product candidates to date across 14 indications, including ulcerative colitis, or UC, Crohn’s disease, or CD, lupus, rheumatoid arthritis, nonalcoholic steatohepatitis, or NASH, multiple sclerosis, Alzheimer’s disease, asthma, psoriasis, atopic dermatitis eosinophilic esophagitis, or EoE, chronic obstructive pulmonary disease, or COPD, diabetic nephropathy and type 1 diabetes. Our initial focus is the development of BT-11 and NX-13 for the treatment of UC and CD.
We expect to commence an integrated Phase 3 trial of BT-11 in UC patients in the United States, Russia, Asia, and Europe in 2021, subject to review of the complete Phase 2 data and FDA feedback, and we expect to discuss with the FDA the pathway for further development of BT-11 in UC patients. We believe the therapeutics we discover and develop, if approved, will have a significant impact on the quality of life of patients suffering from autoimmune diseases.
We have completed a Phase 1a trial of NX-13 in normal healthy volunteers and identified a maximum tolerated dose, or MTD, that was 10-fold greater than the targeted therapeutic dose, without presentation of serious adverse events. Based on these data, we expect to commence a Phase 1b trial of NX-13 in patients with UC in 2021.
Background in autoimmune diseases
Autoimmune diseases generally result from the loss of self-tolerance in the immune system, causing the immune system to attack healthy organs and tissues. This leads to inflammation of the organs and tissues, causing chronic pain and deterioration or destruction of the ability of these organs to function. Current therapies either broadly prevent the immune system from functioning, in the case of corticosteroids, aminosalicylates, or ASA, and immunosuppressants, or systemically block specific molecules that promote inflammation in the case of biologics and JAK inhibitors. While great strides have been made, existing approaches continue to leave unmet patient need, due in part to significant safety concerns.
Our approach
Our mission is to create safe and effective therapeutic candidates to engage targets in areas of unmet medical need where current treatments have limited efficacy and safety and tolerability concerns. To pursue this mission, we are developing novel, disease-specific oral therapeutic candidates that are designed to address the therapeutic gap in the current treatment paradigm for autoimmune diseases. We leverage our AI-based integrated computational and experimental LANCE platform to discover novel therapeutic targets based on prediction and validation of immunometabolic function. Through our LANCE platform, we have identified novel pathways that stand at immunometabolic intersections of multiple well-established immune processes. We believe these pathways have a greater potential to restore the immune tolerance that is lost in patients with autoimmune disease.
We begin our discovery process with an in silico target discovery and drug development platform. The LANCE precision medicine platform analyzes large datasets from autoimmune disease patients to identify novel expression patterns tied to regulatory functions and ranks and prioritizes new immunometabolic targets that are perturbed in human disease and have high potential to serve as selective portals to the coordination of multiple well-categorized downstream responses tied to autoimmune diseases. We then continue evaluation in silico, identifying thousands of targets within the human immune system to evaluate and refine possibilities to maximize efficiency of time and cost. We include multi-scale modeling of immune and disease processes in virtual representation of target tissues for a specific disease to prioritize targets capable of exerting control on the immune response to a level providing meaningful change at the tissue and clinical level. In parallel, targets are filtered for druggability and safety
1
parameters, removing those that are difficult to activate or inhibit or have potential to be linked to toxicity. We prioritize selection of targets with applicable mechanisms to numerous autoimmune and inflammatory diseases while generating predictions for disease-specific potency and optimal pharmacokinetic, or PK, profiles. We then conduct experimental target validation of top ranked targets in cells and in mouse models of disease using loss-of-function and constitutive expression methods. Upon successful target validation, an internal medicinal chemistry program is initiated to optimize preliminary scaffolds into leads. We evaluate lead compounds through a series of in-house robust target engagement, in vitro culture, multiple animal models for diseases of interest, PK evaluation, preliminary dose range finding, or DRF, toxicity studies in rats and translational studies in human patient samples. We have used this strategy to identify, evaluate and advance seven product candidates across three targets spanning numerous autoimmune and related disorders. Prioritization of these product candidates occurs based on unmet clinical need, therapeutic market size and opportunity, preclinical safety and efficacy data, and performance in translational biomarkers.
Optimally identifying novel immunometabolic targets with our proprietary LANCE platform
We believe our LANCE platform may have several key advantages as compared to traditional drug discovery technologies used in autoimmune diseases, including improved discovery and significant target generation potential, ability to modulate multiple critical pathways via a single target, and cost advantage. We have leveraged our platform to identify seven novel immunometabolic targets to date, two of which we are evaluating in clinical trials, LANCL2 and NLRX1. Using our LANCE platform, we expect to identify further novel targets and develop new product candidates for additional indications such as lupus, rheumatoid arthritis, NASH, type 1 diabetes, multiple sclerosis, Alzheimer’s disease, diabetic nephropathy, asthma, psoriasis, EoE and COPD, with the potential to submit one to two investigational new drug applications, or INDs, per year over the next five years, including at least three new INDs in 2021.
Our portfolio
We are leveraging our LANCE platform, our proprietary computational target discovery engine, to design and develop a pipeline of oral small molecule product candidates with differentiated profiles that are the first to target novel immunometabolic mechanisms to address autoimmune diseases. We currently own worldwide development and commercialization rights to each of our product candidates. Since our inception, we successfully cleared four INDs with the United States Food and Drug Administration, or FDA. We expect to submit at least three additional
2
INDs by the end of 2021. Our ability to execute on our strategy of advancing development of our product candidates will depend on the results of our ongoing and planned clinical trials.

|
* |
Subject to feedback from the FDA, we intend to initially submit one IND for each product candidate in order to initiate Phase 1 clinical trials in an indication. We will submit subsequent INDs to proceed to Phase 1 clinical trials in additional indications for that product candidate. |
|
** |
We define the discovery stage of development as the design, initial synthesis, target binding and preliminary pharmacology testing in in vitro and animal models of disease; the preclinical stage of development as GLP-toxicity and pharmacology studies, metabolism and pharmacokinetics in addition to scale-up chemistry manufacturing and controls for drug substance and drug product; the Phase 1 stage as clinical trials designed to assess safety, tolerability and pharmacokinetics in healthy volunteers or small patient cohorts; the Phase 2 stage as placebo-controlled clinical trials designed to provide proof-of-concept efficacy in the target patient population; and the Phase 3 stage as clinical trials designed to provide the final supporting data for the safety and effectiveness of a drug in the proposed indication to support an NDA filing. |
Numerous anticipated milestones in our portfolio are currently in preparation. For BT-11, a Phase 3 clinical trial in UC is expected to be initiated in the second half of 2021, subject to review of the complete Phase 2 data and FDA feedback, a Phase 2 clinical trial of BT-11 in CD will be initiated in the first half of 2021, an IND filing of BT-11 in EoE has reached FDA clearance, and IND filings of BT-11 in psoriasis and atopic dermatitis will occur in 2021. For NX-13, a Phase 1b clinical trial in UC is expected to be initiated in the first half of 2021 and a Phase 2 clinical trial in CD is expected to be initiated in 2022, respectively. For BT-104, IND-enabling studies are expected to be completed in 2021. For PX-69, IND-enabling studies are expected to be completed in the first half of 2022.
3
The successful clinical development of our product candidates is dependent on many factors beyond our control, including timely and successful completion of preclinical studies and clinical trials required to progress our clinical programs, and the sufficiency of our financial and other internal and external resources to complete such studies and trials, file INDs and NDAs, as appropriate, and commercialize BT-11 for UC, effective INDs from the FDA or comparable foreign applications that allow commencement of our planned clinical trials for discovery or preclinical-stage product candidates, receipt of timely marketing approvals from the FDA or other applicable regulatory authorities, our ability to successfully launch commercial sales of any approved product candidates, whether independently or with partners and our ability to produce and approved product candidates on a commercial scale. See “Risk factors—Risks related to the discovery, development and commercialization of our product candidates,” “Risk factors—Risks related to our dependence on third parties” and “Management’s discussion and analysis of financial condition and results of operations - Liquidity and capital resources.”
BT-11 program overview
Our lead product candidate, BT-11, a gut-restricted oral therapeutic that is the first candidate designed to engage the novel target lanthionine synthetase C-like protein 2, or LANCL2, a membrane receptor that has been shown to modulate immunological mechanisms that are associated with autoimmune diseases such as UC or CD. Approximately 2 million Americans suffer from UC or CD, and the treatment of these two diseases in the United States accounts for an aggregate annual market size of up to $17 billion. There are currently no approved treatments targeting LANCL2. BT-11 is designed to act locally in the gastrointestinal tract for treatment of inflammatory bowel disease, or IBD. We have designed BT-11 to overcome limitations of existing therapies, including with respect to side effect profile, route of administration (injectables) and sustained efficacy.
We have completed the induction phase of a Phase 2 clinical trial of BT-11 for mild to moderate UC in the United States, Russia and Europe. We expect to commence an integrated Phase 3 trial of BT-11 in UC patients in the United States, Russia, Asia, and Europe in 2021, subject to review of the complete Phase 2 data and FDA feedback, and we expect to discuss with the FDA the pathway for further development of BT-11 in UC patients. We also expect to initiate a Phase 2 trial of BT-11 for the treatment of moderate to severe CD in the first half of 2021. We believe that BT-11, if approved, has the potential to disrupt the treatment paradigm in IBD. In UC, we believe BT-11 has the potential to be utilized as a pre-biologic therapy for mild-to-moderate cases, where 70% to 80% of the patient population exists, due to its novel mechanism of action and tolerability profile, with no dose-limiting toxicities observed in preclinical studies in doses up to 7-fold higher than the proposed clinical therapeutic dose. In CD, we believe that BT-11, if approved, has the potential to serve as a first-line therapy and treat patients prior to their progression into biologics by addressing the main limitations of current therapeutics. We believe that an oral, gut-restricted small molecule delivered once daily in a singular tablet could serve as a first-line therapy in the moderate to severe CD treatment paradigm.
NX-13 program overview
Our second product candidate, NX-13, is a novel, gut-restricted oral therapeutic that targets NOD-like receptor X1, or NLRX1, a mitochondria-associated receptor that has been associated with the modulation of inflammatory cytokines for UC and CD. There are currently no approved NLRX1-based drugs. NX-13 is designed to target NLRX1 in order to induce anti-inflammatory effects in CD4+ T cells and other immune cells in the gastrointestinal tract. We believe that, if approved, NX-13 could provide an additional treatment option for the up to 50% of UC patients that experience relapse within one year of current therapies and up to 55% of CD patients that relapse within one year of achieving active remission. Mechanistically, NX-13 could provide improvements in fibrosis. We believe NX-13 offers the potential, both as a single agent and in combination with other therapeutics, to address the unmet medical need in UC and CD. We have completed a Phase 1a trial of NX-13 in normal healthy volunteers and identified a MTD that was 10-fold greater than the targeted therapeutic dose, without presentation of serious adverse events. Based on these data, we expect to commence a Phase 1b trial of NX-13 in patients with UC in 2021.
Autoimmune market opportunity
We are developing therapeutics for the large and growing autoimmune disease market, which is expected to reach $153 billion by 2025. Our lead product candidates target a multi-billion dollar market opportunity across 13 different indications.
4
The UC and CD markets are currently dominated by biologic drugs that generated up to $17 billion in U.S. sales in 2019. From 2010 to 2020, the IBD market experienced a compound annual growth rate of 13.1%. Approximately 35% of patients treated with biologics fail to enter remission. Meanwhile, greater than 90% of UC patients with active disease have mild to moderate disease and 50% to 60% of IBD patients are biologic naïve. Current market competition in this space is limited to aminosalicylates, steroids and immunosuppressants which have only low efficacy in induction and maintenance of clinical remission. The primary competition in development, falling into two primary classes (JAK inhibitors, S1P modulators), targets a moderate to severe patient population that is primarily comprised of patients who have not responded to biologics, due to inherent safety concerns with these classes. With the exception of Xeljanz, which is FDA-approved for the treatment of UC, oral agents approved for IBD are limited to aminosalicylates, steroids and immunosuppressants that have limited efficacy. We believe we have the potential to address the large IBD market and specific therapeutic gaps in both UC and CD. However, there is no guarantee that any of our product candidates will be approved by the FDA and, even if approved, there is no guarantee that our product candidates will earn revenues comparable to Entyvio, Humira or Stelara. The graphic and table below depict the large market opportunity in IBD.

EvaluatePharma® May 2020, Evaluate Ltd.
5
Large market opportunities for product candidates targeting IBD

EvaluatePharma® August 2020, Evaluate Ltd.
Our strengths
We believe we are a leader in the discovery of targets through novel multi-modal pathways at the interface of immunity and metabolism. Through our proprietary LANCE precision medicine platform, we have pioneered a promising drug development engine underpinned by advanced computational capabilities applied to the discovery of new therapeutic targets for autoimmune disease.
Our distinctive strengths include:
|
• |
We are pioneering a new treatment paradigm in immunometabolism based on novel pathways. Historically, innovation in autoimmune drug discovery has focused on the same known targets creating new molecules whose development has been hampered by toxicity issues connected to the mechanism of action or systemic pathways and which have provided only marginal change to efficacy at best. The late-stage development pipeline in IBD can be summarized by JAK/STAT-, cytokine- and adhesion-based mechanisms of action, while our lead product candidate, BT-11, is the most advanced immunometabolic asset in clinical development. Our unique ability to identify novel targets that intersect and provide upstream control of established inflammatory and regulatory pathways supports a clinical path to develop novel drugs. We are further leveraging these novel immunometabolic targets in a disease-specific way with an aim to provide better and more precise care to patients. |
|
• |
We have discovered and are developing several clinical-stage assets across the LANCL2 and NLRX1 pathways through our LANCE platform and distinctive capabilities. To date, we have identified seven novel immunometabolic targets, of which candidates that target two (LANCL2 and NLRX1) have progressed into the clinic. Our lead asset, BT-11, is an orally-active, gut-restricted, small molecule that is the first therapeutic candidate to target the LANCL2 pathway. We have completed the induction phase of a Phase 2 clinical trial of BT-11 for mild to moderate UC in the United States, Russia and Europe. Based on this trial data, we expect to commence an integrated Phase 3 trial of BT-11 in UC patients in the United States, Russia, Asia, and Europe in the second half of 2021, subject to review of the complete Phase 2 data and FDA feedback, and we expect to discuss with the FDA the pathway for further development of BT-11 in UC patients. We also expect to initiate a Phase 2 trial of BT-11 for the treatment of moderate to severe CD in the first half of 2021. Our second asset, NX-13, targets the NLRX1 pathway and has completed Phase 1a clinical testing without presentation of serious adverse events and a MTD that was 10-fold greater than the targeted therapeutic dose. Based on these data, we expect to commence a Phase 1b trial of NX-13 in patients with UC in 2021. |
|
• |
We are developing product candidates with novel mechanisms of action and the potential for use as monotherapy or combination therapy to address therapeutic gaps for IBD patients where current treatments have limited efficacy and safety and tolerability concerns. We believe the LANCL2 pathway is not targeted by any other drug in development for IBD. With a dual effect for pro-regulatory and anti-inflammatory signaling, |
6
|
targeting this pathway may offer a unique advantage for longer maintenance of clinical remission, decreasing the high flare rates in IBD. We believe BT-11, if approved, has the potential to disrupt the treatment paradigm in UC as a pre-biologic therapy for mild-to-moderate cases, where greater than 90% of the patient population with active disease exists, due to its novel mechanism of action and no dose-limiting toxicities in doses up to 7-fold higher than the currently proposed therapeutic dose in our completed Phase 1 clinical trial. Additionally, BT-11 and NX-13 are designed to offer a convenient, once-daily oral dose which we believe may provide competitive advantage over the injectable, systemically distributed biologics. Given the novel targets and limited toxicities, we believe BT-11 and NX-13 have potential to be used as monotherapy or in combination with other therapies in IBD. |
|
• |
We are targeting several indications characterized by unmet medical need and therapeutic gaps that represent a broad market opportunity. We are developing therapeutics for the large and growing autoimmune disease market, which is expected to reach $153 billion by 2025. Our lead product candidates target a multi-billion dollar market opportunity across 13 different indications. The global UC market was $5.3 billion in 2016, and is expected to grow at a 2.5% compound annual growth rate between 2016 and 2026. The global CD market is estimated at approximately $12 billion and expected to grow at 4% compound annual growth rate between 2019 and 2022. We believe these indications provide a large market opportunity with no established satisfactory standard of care. The primary competition in development targets a moderate-to-severe patient population that is primarily comprised of patients refractory to biologics, due to inherent safety concerns within these classes. Many key opinion leaders expect oral agents to expand the overall autoimmune disease therapeutic market size and accelerate growth in the IBD space. We believe our product candidates, if approved, have the potential to address the large autoimmune disease therapeutic market and specific therapeutic gaps in both UC and CD. Our broad therapeutic pipeline is comprised of potential valuable treatments for other autoimmune diseases, such as lupus, rheumatoid arthritis, multiple sclerosis, and type 1 diabetes, as well as other indications with immunometabolic components, such as NASH, Alzheimer’s disease, and asthma. |
|
• |
We have a strong intellectual property foundation and we retain global commercial rights to all of our product candidates. Our intellectual property portfolio includes composition of matter and method of use patents covering both BT-11 and NX-13. Our lead product candidates’ patent protection extends until 2035, in the case of BT-11, and 2039, in the case of NX-13. Our intellectual property portfolio includes more than 75 issued patents and patent applications. Furthermore, we have retained exclusive development and commercial rights to all of our product candidates. |
|
• |
We have assembled an experienced team comprised of industry leaders in the fields of immunology and inflammation and we are backed by prominent life science investors. Our management team has broad expertise and successful track records in drug discovery, immunology & inflammation, clinical development, regulatory affairs, manufacturing and commercialization of therapies, as well as in business and finance, through previous experiences at leading institutions. We are also guided by our board of directors, and a clinical advisory board composed of key opinion leaders in CD and UC. |
Our strategy
As a leader in the development of therapeutic candidates addressing immunometabolic mechanisms of action, we believe we are distinctly positioned to advance the discovery and development of potentially safer and more effective novel therapeutics for autoimmune diseases. We are targeting indications characterized by high unmet clinical need, significant likelihood of success in clinical development and the ability to improve the quality of life for large patient populations. Our strategy consists of the following key components:
|
• |
Maximize and capture value in our LANCE platform by discovering and developing oral therapeutics that are the first to target novel immunometabolic pathways to improve the lives of patients with autoimmune diseases. We have leveraged our experience in immunology and therapeutic development, underpinned by the guidance of our clinical advisory board, to build the LANCE platform. We will apply our proprietary artificial intelligence, or AI, and advanced computational modeling capabilities to continue to identify novel therapeutic targets based on predictions of immunometabolic function. Through our LANCE platform, we have progressed BT-11 from initial discovery to an ongoing Phase 2 clinical trial in less than three years, and in a capital efficient manner. In addition to LANCL2, NLRX1, and PLXDC2, the LANCE platform has identified four new targets in early preclinical stages of development. We have led the discovery of the immunometabolic mechanisms for these three targets independently of external research and possess the capacity to internally discover novel targets. We intend to continue to leverage the LANCE platform to further advance a pipeline of product candidates for the treatment of autoimmune diseases. The LANCE platform has served and, we believe, will continue to serve in the generation of a seamless therapeutic pipeline ranging from early preclinical to lead selection to clinical development and commercialization. |
7
|
• |
Advance the development of our lead product candidate, BT-11, into Phase 3 clinical trials for the treatment of UC and CD. We identified LANCL2 as a critical autoimmune pathway for multiple IBD conditions and have designed BT-11 to overcome limitations of existing therapies including with respect to side effect profile, route of administration, efficacy and bridging the therapeutic gap between ASA failures and biologics. We expect to commence an integrated Phase 3 trial of BT-11 in UC patients in the United States, Russia, Asia, and Europe in the second half of 2021, subject to review of the complete Phase 2 data and FDA feedback, and we expect to discuss with the FDA the pathway for further development of BT-11 in UC patients. We also expect to initiate a Phase 2 trial of BT-11 for the treatment of moderate to severe CD in the first half of 2021. |
|
• |
Advance our second clinical candidate, NX-13, into Phase 1b/2 clinical trials for the treatment of UC and CD. NLRX1 is a novel target at the intersection of the pathogenesis of IBD with the ability to modulate multiple associated complications including deficiencies in the epithelial barrier, interactions with the gut microbiome and underlying inflammation. We have completed a Phase 1a clinical testing of NX-13, and we expect to commence a Phase 1b trial of NX-13 in patients with UC in 2021. We intend to explore NX-13 both as a monotherapy and in combination with other therapeutics for the treatment of UC and CD. |
|
• |
Continue to develop a franchise of oral therapies from our deep preclinical pipeline of product candidates targeting immunometabolic pathways identified by our LANCE platform. Recognizing the applicability of our novel targets across a range of autoimmune conditions, and the benefits to patients of disease specific molecules, we intend to advance each of our preclinical assets to exploit the full potential of each pathway or target. We are building upon our precision medicine approach to develop disease-specific molecules by exploring the use of biomarkers to identify patients more likely to benefit from therapies. BT-104, BT-111, NX-66, NX-73 and PX-69 have been identified as possible valuable treatments for autoimmune diseases, such as lupus, rheumatoid arthritis, multiple sclerosis, and type 1 diabetes, as well as other indications with immunometabolic components, such as NASH, Alzheimer’s disease, and asthma. We expect to file at least one to two INDs per year over the next five years, including three new INDs in 2021. |
|
• |
Maximize the commercial value of our therapeutic pipeline. We have retained worldwide commercial rights to all of our therapeutic programs. We intend to continue advancing more clinical candidates in various therapeutic areas independently. We plan to become a fully integrated pharmaceutical company by selecting promising programs in specialty pharmaceutical and orphan indications for internal development and commercialization and developing a commercial arm to capture value for shareholders. We will also pursue territory deals that enable partnering on commercialization of lead therapeutic assets outside of the U.S. and European markets. Moreover, we will consider partnering with strategics to develop some of the follow on therapeutic assets as a means of monetizing some of our pipeline assets. |
Background on immunometabolism
Alterations in the concentration of metabolic enzymes and substrates in the cell often precede changes in the expression of genes that articulate inflammatory responses in autoimmune diseases. These metabolic processes are critical determinants of the function of immune cells. In the course of autoimmune diseases, multiple receptors transmit information about the environment through pathways at the intersection of immunity and metabolism. Over the past decade, this field of study, known as immunometabolism, has revealed the complex metabolic pathways that underlie autoimmune response in disease and has allowed us to focus on developing ways to manipulate these regulatory networks to enhance and control immunity.
Genes with inflammatory functions, such as TNF or IL-6, tend to be overexpressed during autoimmune responses and are easily identified. The metabolic pathways of immune cells must also be configured to meet the demands of their function. For example, the pro-inflammatory subset of immune cells known as T-effector cells (Th1 and Th17) depend upon glycolysis and oxidative phosphorylation, while cells involved in anti-inflammatory response known as regulatory T-cells utilize predominately fatty acid oxidation. However, genes with immunometabolic roles experience modest change in expression levels though they are critical in modulating inflammation. By altering the signals that drive differentiation and the metabolic pathways that support it, immune tolerance can be reestablished in patients where there is immune dysregulation, such as in autoimmune disease.
We are focused on developing therapeutic interventions based on activating molecular targets within these immunometabolic pathways to correct aberrant inflammatory responses. We have developed an AI platform that allows for efficient identification, segregation and prioritization of these genes from high throughput datasets. This platform is designed to differentiate our program of target discovery and product development.
8
Our LANCE platform
We have built our LANCE precision medicine platform to identify novel targets at the intersection of immunity and metabolism and to develop new therapeutic products for these new targets. Our innovative approach to immunometabolism applies proprietary AI and advanced computational modeling to identify novel therapeutic targets based on predictions of immunometabolic function and create therapeutics to engage those targets in areas of unmet medical need. Upon target discovery, we have efficient medicinal chemistry and preclinical capabilities, as evidenced by the less than 24 months from discovery to Phase 1 initiation with BT-11 and less than 18 months from discovery to Phase 1 initiation with NX-13. Our development time from discovery to Phase 2 ready was 30 months, in the case of BT-11, and 24 months, in the case of NX-13. Using our LANCE platform, we expect to submit one to two INDs per year over the next five years, including at least three INDs in 2021. We believe similar timelines are possible with future product candidates; however, the process of clinical development is inherently uncertain and no guarantee can be made that similar timelines will be achieved.
We have successfully leveraged this iterative process to identify seven novel immunometabolic targets to date, and product candidates which are designed to target two such targets, LANCL2 and NLRX1, have begun clinical trials pursuant to INDs. Using our LANCE platform, we not only expect to identify further novel targets but to also continue to develop disease-specific product candidates for indications such as lupus, rheumatoid arthritis, NASH, type 1 diabetes, multiple sclerosis, EoE, psoriasis, Alzheimer’s disease, diabetic nephropathy, asthma and COPD. We believe our innovative precision medicine approach, in which we develop specific products for disease states, enhances our ability to address existing unmet need for patients.
We developed our proprietary LANCE platform to revolutionize the target identification, prioritization and selection process as well as facilitate the development of new therapeutic products for those targets. Our leadership team has deep experience in this field, including Dr. Bassaganya-Riera, a pioneer in computational immunology and precision medicine. The team has published over 30 computationally- and AI-focused peer-reviewed publications, such as in Artificial Intelligence in Medicine and PLOS Computational Biology, and has created foundational large-scale models of the immune system, including one of the most widely used ordinary differential equation-based model of CD4+ T cell, or Treg, differentiation. The LANCE platform embodies the re-focusing of this know-how into the application of drug development for autoimmune disease.
Processes of our platform
The LANCE platform is an innovation engine that uses four main processes to identify novel therapeutic immunometabolic pathways with significant potential to exert immunoregulatory control of autoimmune disease:
|
• |
Data extraction. We extract data from both proprietary and public omics datasets from human diseases with defined categorical data tied to responders and non-responders, active disease versus remission, and trends and signature patterns over the progression of disease. The output is a disease-agnostic composite database of autoimmune diseases with over 10,000 potential targets. We continually update this database, resulting in new opportunities to identify pathways and the ability to enhance the robustness of previous predictions. |
|
• |
Computational target analysis. We use proprietary AI-based approaches to analyze our database, compiling and inferring hidden blind spots and linkages in signaling networks that may serve as understudied, yet potent, portals into modulating immunometabolism. Our blind spot algorithm specializes in identifying the targets for which the connections to known downstream functional events were previously undiscovered. The output is a ranked priority list of 80 to 100 targets based on relative impact on prominent immune and metabolic pathways. Based on the weighting of individual pathways and subsetting of data analytical methods, the system is versatile enough to offer cross-validation potential and continuous mining of the high-dimensional data. |
|
• |
In silico immune system modeling. We perform multiscale modeling of the immune system to link molecular signaling to changes in overall tissue and patient level responses to prioritize targets within a specific disease. The output is narrowed down to five to ten targets identified to have significant in silico impact on intricately calibrated models of human autoimmune disease that capture complex interactions between molecules and cells in disease-specific target tissues. This encompasses the colon for UC and CD, the entire gastrointestinal tract for CD, synovium for rheumatoid arthritis, lung for asthma and COPD, brain for multiple sclerosis and Alzheimer’s disease, pancreas for type 1 diabetes, liver and adipose tissue for NASH, and germinal centers and sites of immune complex reactions in spleen, kidney, circulation and a variety of other organs for lupus, and kidney for diabetic nephropathy. |
9
|
• |
Preclinical validation and prioritization. We characterize and validate immune responses using in vitro and in vivo systems. We prioritize targets based on: |
|
|
• |
Preclinical efficacy: We utilize two or more independent animal models, primary patient cells and organotypic systems, as well as flow cytometry, gene expression, histology and metabolic tests, to evaluate preclinical efficacy. |
|
|
• |
Tolerability: We strive for a no-observed-adverse-effect-level, or NOAEL, at 50 times the anticipated clinical therapeutic dose using mouse, rat and pig models. |
|
|
• |
Biomarkers: We prioritize targets where there are available translational biomarkers to make early preclinical and clinical go/no-go decisions. |
|
|
• |
Indication: We prioritize targets that have a significant market size and a greater potential for success in clinical trials. |
|
|
• |
Competition: We seek targets where there is an unmet clinical need in the therapeutic indication. |
The result is selection of two to three targets preclinically validated to impact disease pathogenesis, for which medicinal chemistry programs can be established. The graphic below summarizes the selection process of our LANCE platform.

Foundations of the LANCE platform
Across autoimmune disease, the current therapeutic strategy is often the same: block a single cytokine, use a broadly immunosuppressive steroid and alter lymphocyte trafficking. We believe this paradigm fails to effectively modulate the massively and dynamically interacting immunological network to provide sustained therapeutic responses during autoimmune disease. As a result, patients that are non-responsive to one class of drugs have a low likelihood to respond to other classes due to the similarity between classes. In addition, despite chronic treatment with immunosuppressive drugs, relapse and flare rates in all autoimmune diseases are high. In UC and CD, half of patients relapse within a year of starting treatment. In rheumatoid arthritis, 90% or more fail to achieve extended remission. In systemic lupus erythematosus, or SLE, and multiple sclerosis, frequent steroid regimens are needed to address flares.
10
To address the limitations of current therapies for autoimmune diseases, we utilize our LANCE platform to develop therapeutics based on the following foundational pillars:
Targeting immunometabolism. An abundance of recent evidence supports that the function and phenotype of immune cells, inclusive of both innate and adaptive immune response, is highly conserved and intertwined with metabolic processes. The synthesis of building blocks for cytokines, antibodies and lipid mediators, the preparation for proliferation, the processes of phagocytosis and autophagy, and the generation of long-enduring memory cells require differential metabolism. The therapeutic manipulation of cellular metabolism is a robust multimodal mechanism to shift the balance between effector and regulatory immune cells. These strategies have resulted in resounding successes in the fields of immuno-oncology (such as pembrolizumab, an anti-PD1, ritonavir, a protease inhibitor, and everolimus, an mTOR inhibitor) and metabolism (such as metformin, an antidiabetic, and statins, lipid-lowering drugs) but remain a yet unexploited opportunity in autoimmune disease. With a multitude of genetic instability characterizing autoimmune disease patients, therapeutics targeting well-conserved and stable immunometabolic processes could prove a successful strategy for therapeutic efficacy.
Prioritizing patient safety. We have prioritized the development of product candidates targeting LANCL2 and NLRX1 based not only on potent anti-inflammatory effects observed in preclinical studies but also on the observed ability to modulate their signaling without dose-limiting toxicities in completed nonclinical and clinical studies. For BT-11, NX-13 and other product candidates in our pipeline, the primary mechanism of action is designed to result in the activation of immunoregulatory effects, as opposed to complete inhibition of natural immunological processes. As a result, our product candidates are designed to result in a functional immune system in which self-tolerance and immune homeostasis are restored. For the majority of autoimmune diseases, including UC and CD, safety concerns are a primary cause of gaps in the treatment paradigm between patients with mild disease severity and patients treated with biologics and other treatment classes with “black-box” label safety warnings.
Restoring immune tolerance. We view the immune system as a massively and dynamically interacting complex system. By inhibiting single cytokines and narrow pathways, multiple compensatory mechanisms can emerge, making any therapeutic improvement in disease short-lived. Each of our lead targets is designed to induce regulatory mechanisms that are conserved, meaning the mechanisms have specific effects on intended pathways. These mechanisms are multi-pronged and are tied to contact-mediated suppression, production of anti-inflammatory cytokines, such as IL-10, and metabolic changes favorable to Tregs; all of which may drive more durable clinical response and remission.
Identifying novel pathways. A primary objective of our LANCE platform is to identify novel pathways with conserved multi-pronged mechanisms with the potential to elicit effects beyond those observed in current classes of drugs targeting autoimmune disease. In particular, our process focuses on identifying central nodes in the network which emanate multiple spokes to coordinate downstream responses. For example, the activation of LANCL2 interacts with CD25/STAT5 signaling, decreases late-stage glycolysis while enhancing oxidative phosphorylation, and influences secondary messenger signaling. Based on our robust dataset, we believe this coordination will enhance the robustness of response to our product candidates through built-in redundancies capable of maintaining coordination should one spoke be rendered non-functional due to a disease-associated mutation in a specific patient. Despite the novelty of the target and certain downstream pathways, our validation is rooted in well-categorized biomarkers of inflammation, including decreases in TNF expression, NF-κB activity, or fecal calprotectin levels with the activation of LANCL2 and NLRX1. We believe elucidation of these well-validated downstream anchors increases the confidence for the translation of preclinical results when targeting our novel pathways. To date, we
11
have identified seven novel pathways using the LANCE platform with promising potential to innovate the treatment of autoimmune disease.

Benefits of our LANCE platform
We believe our LANCE platform has several key advantages, including:
|
• |
Improved drug discovery paradigm in autoimmune diseases: Our LANCE platform is designed to achieve an efficient discovery timeline, from target identification to lead compound in under 12 months. Our seven identified candidates have moved to in vitro and in vivo studies without a single discontinuation for observations related to toxicity, efficacy or non-druggability. Finally, customizable in silico analysis to identify targets agnostically as overall regulators of immunology or focused on a particular indication of interest broadens the breadth of our opportunities to a series of new potential indications. |
|
• |
Modulating multiple critical pathways via a single target: The LANCE platform provides the potential ability to identify candidates which could coordinate multiple cellular processes into the same overall phenotype to reduce compensatory mechanisms tied to loss of response. The LANCE platform provides robust, well-conserved potential targets in an autoimmune disease space associated with high patient-to-patient variability. The ability to modulate multiple primary targets and several escape mechanisms offers the potential to identify candidates which could be developed to provide a more durable effect of therapy. |
|
• |
Cost advantage and meaningful target generation potential: Given the demonstrated capital efficiency of the initial drug development programs we have undertaken to date, we believe our LANCE platform is likely to continue providing for a decrease in discovery and development costs relative to licensing new product candidates as we advance the timeline of discovery to IND. We also believe the LANCE platform may provide meaningful potential to discover new targets and re-prioritize identified targets with continually evolving input data. Streamlined target selection and prioritized list of selected targets may also minimize time and cost dedicated to discovery stage. A rational and efficient product prioritization from the large targets pool offered by our platform is bolstered by efficacy studies with two or more animal models, by toxicity and PK data with target tissue engagement in mice, rats and pigs, as well as by the availability of translational biomarkers to make early preclinical and clinical go/no-go decisions. We are targeting indications characterized by high unmet clinical need, significant likelihood of success in clinical development and market size opportunity. |
12
Our LANCL2 pathway product candidates
The LANCL2 pathway
The LANCL2 receptor triggers a unique reaction at the interface of immunity and metabolism. LANCL2 is a surface membrane-associated receptor and one of three members of the LANCL family, which have been previously studied in detail. Our analysis with the LANCE platform has produced a deeper understanding of the role of the LANCL2 pathway in modulating key cellular and molecular genes tied to autoimmune disease. More specifically, activation of LANCL2 is designed to intercept autoimmune disease at multiple levels through extensively-characterized pathways and signaling molecules, including decrease of inflammation-promoting TNFα, IFNγ, IL-6 and MCP1 and increase in regulatory Tregs anti-inflammatory activities.
Given the differential metabolic preferences of Th1 and Th17 versus Treg cells, whereby the former prefer glycolytic pathways versus the latter prefer oxidative phosphorylation to produce energy, the rewiring of metabolism between oxidative and glycolytic metabolic pathways mediated by LANCL2 activation is designed to produce a functional switch in immune cells that promotes regulatory functions. For instance, highly proliferative effector cells associated with autoimmune reactions produce strongly inflammatory cytokines, such as TNFα and IFNγ, and differentiate into effector Th1 and Th17 CD4+ T cell subsets. Both of these events are associated with lactate, the key metabolite of anaerobic glycolysis. In contrast, when LANCL2 is activated, it promotes the function of enzymes that diminish lactate and favor oxidative phosphorylation. This metabolic switch is supportive of stable expression of FOXP3, the master transcription factor associated with the function of Treg cells.
In inflammatory tissue microenvironments tied to autoimmune disease, loss of CD25 (the IL-2 receptor) is one of the first changes in Tregs to occur, leading to co-production of inflammatory cytokines and an overall weakened suppressive potential. LANCL2 activation provides protection from and restoration of IL-2 signaling by augmenting STAT5 phosphorylation. As a result, LANCL2 activation by BT-11 and other product candidates that target LANCL2 results in stable Tregs that have enhanced suppressive capacity as evidenced by expression of key regulatory molecules such as immune checkpoint inhibitors like PD1. The importance of LANCL2 in Tregs has been independently validated in a proteomics-based screen identifying differential expression of LANCL2 in Tregs compared to non-Treg cells.
The graphics below illustrate key elements of LANCL2 activation by BT-11.
|
•BT-11 Multimodal MoA: •Decreases the production of inflammatory mediators tied to IBD (TNFα, IFNγ, MCP1, IL-6, and IL-8) •Increases anti-inflammatory molecules in Tregs that protect from autoimmunity (IL-10, FOXP3) •BT-11 generates suppressive regulatory CD4+ T cells (Tregs) that restore and maintain immune tolerance in the Gl tract •Decrease proliferation and differentiation of effector CD4+ T cells (Th1 and Th17) •Aid in epithelial cell and support the reduction of IL-8 and chemokine-dependent neutrophil influx Leber, A., et al. J lmmunol, 2019. |
|

13
|
•Multipronged mechanism of action targeting downstream known immunological targets tied to autoimmune diseases, including IBD. •BT-11 enhances CD25/STAT5 signaling to support the stable differentiation of regulatory CD4+ T cells with greater anti-inflammatory functionality. •BT-11 increases PDH activity, resulting in increased oxidative metabolism supporting FOXP3 stability. •BT-11 downregulates glycolytic pathways associated with TNF-α production and effector CD4+ T cells, including production of lactate and over-expression of EN01. •BT-11 increases suppressive effects of Tregs due to enhanced immune checkpoint surface markers (LAG3 and PD-1). |
|

While critical to functions in Tregs, the LANCL2 receptor is expressed in a wide range of immune cells, epithelial cells and cells from metabolic (muscle, adipose and liver) tissues. The effects within these cell types often mirror the described signaling events within Tregs, whereby the promotion of mitochondrial metabolism parallels potent anti-inflammatory markers. In IBD, mitochondrial metabolism pathways account for the majority of downregulated genes relative to healthy controls. A primary cause of this downregulation is a direct result of defective regulation of mitochondrial metabolism in intestinal epithelial cells. In both mouse models of IBD and intestinal epithelial organoids ex vivo, activation of mitochondrial function has been observed with LANCL2 activation, restoring this metabolism to healthy levels. When this restoration is achieved, chemokine production from intestinal epithelial cells is decreased, leading to suppression of neutrophil recruitment into the intestinal wall. In IBD, neutrophils are crucial histological markers of active disease as well as the primary source for calprotectin, a highly predictive fecal biomarker of response to treatment since the majority of drugs approved for treating IBD cause a drop in fecal calprotectin concentrations. Neutrophils and fecal calprotectin were significantly decreased by LANCL2 activation in all preclinical models tested and in our Phase 1 clinical trial, fecal calprotectin concentrations were also decreased following BT-11 treatment. In phagocytes (macrophages and dendritic cells), LANCL2 activation of mitochondrial metabolism supports the efficient processing of harmless cellular material from the body taken up by these cells leading to increased production of IL-10 from these cells and lower auto-reactivity to self-antigens. In metabolic tissues, LANCL2 activation results in increased AMPK signaling, which increases mitochondrial function and synergizes with insulin to activate efficient energy storage in both muscle and liver.
With robust control of both immune and metabolic signaling, we believe the novel LANCL2 pathway is relevant in a wide range of autoimmune, inflammatory and metabolic diseases, specifically CD, UC, rheumatoid arthritis, lupus, NASH, type 1 diabetes and psoriasis. We have prioritized IBD as our first indication in the LANCL2 pathway due to the significant unmet clinical need and major therapeutic gaps in both UC and CD and the mechanistic relevance in both Tregs and epithelial cell metabolism. Validation of the LANCL2 pathway in IBD with BT-11 may have wide-reaching implications on the development of new treatments for other diseases. For example, increased Treg stability and IL-10 production from phagocytes may be critical in restoring self-tolerance in systemic lupus erythematosus. In addition, shifting the balance between Treg and Th17 cells and reducing neutrophil recruitment are highly relevant to improvement of symptoms in rheumatoid arthritis. Correcting the underlying deficiencies in insulin sensitivity and reducing chronic low-grade systemic inflammation has potential to improve NASH. Decreasing Th2 responses might provide a therapeutic benefit in EoE. Modulating T cell responses and TNF in the skin might provide a therapeutic benefit in psoriasis. We are initially focused on developing three product candidates that target the LANCL2 pathway: BT-11, BT-104 and BT-111.
14
BT-11, an oral LANCL2 agonist for the treatment of ulcerative colitis and Crohn’s disease
Overview
BT-11 is an oral, gut-restricted, small molecule that is the first product candidate to target LANCL2 which we are evaluating in clinical trials in both UC and CD. UC and CD are chronic autoimmune diseases with significant therapeutic gaps resulting from safety concerns and modest efficacy of current treatments. We believe that an oral, gut-restricted small molecule delivered once daily in a singular tablet could address the therapeutic gaps in the UC and CD treatment paradigms and have a significant impact on quality of life for IBD patients. BT-11 is a wholly-owned product candidate that has successfully completed a Phase 1 clinical trial. We have completed the induction phase of a Phase 2 clinical trial of BT-11 for mild to moderate UC in the United States, Russia and Europe. Based on the data from this trial, we expect to commence a Phase 3 trial of BT-11 in mild to moderate UC patients in the United States, Russia, Asia, and Europe in the second half of 2021, subject to review of the complete Phase 2 data and FDA feedback, and we expect to discuss with the FDA the pathway for further development of BT-11 in UC patients. We expect to commence a Phase 2 proof-of-concept study in patients with moderate to severe CD in the first half of 2021. We have successfully filed a new IND for a new orodispersable BT-11 formulation for EoE in March of 2021.
Background on UC and current treatments
UC is a chronic, autoimmune, inflammatory bowel disease that causes inflammation, irritation, and ulcers in the lining of the large intestine (colon) and rectum. Symptoms include abdominal pain, rectal pain and bleeding, bloody stools, diarrhea, fever, weight loss, and malnutrition. Having UC puts a patient at increased risk of developing colon cancer. Diagnosis typically occurs in early adulthood and the disease requires maintenance treatment for the remainder of the patient’s life. UC is estimated to affect over 900,000 patients in the United States and over 1 million patients throughout the rest of the world. The global therapeutics market for UC was $5.3 billion in 2016 and is expected to grow at a 2.5% compound annual growth rate between 2016 and 2026. Of this global market, the United States market comprised over $4 billion in 2019. With 70% of addressable patients experiencing a second flare within one year and 30% of patients in remission failing to stay in remission for more than one year, there is an unmet medical need in UC for safer and more efficacious therapeutics.

Source: GlobalData Market Research Report
15
Patients with UC are classified into mild-to-moderate, comprising 70-80% of patients, and moderate-to-severe, based on the level of symptoms experienced. Accordingly, the current therapeutic treatments for UC depend on the severity of the disease and are broadly divided into five classes:
Mild to moderate UC
The following treatments are typically used in the treatment of mild to moderate UC:
|
• |
Aminosalicylates or 5-ASA (mesalamine, sulfasalazine) are used as a first-line therapy in UC without a precise mechanism of action. Approximately 80% of patients qualifying as mild to moderate in severity receive mesalamine as a maintenance drug, but many require corticosteroids to address disease flares and eventually lose response progressing into immunosuppressants or biologics. Aminosalicylates are commonly preferred by physicians and patients for the treatment of IBD, due to high tolerability in most patients. Common side effects include headache, nausea, dyspepsia, flatulence and diarrhea. Rare but more serious side effects include pleuritis, pericarditis, myocarditis, pancreatitis, cholestic hepatitis, nephritis and renal dysfunction. |
|
• |
Corticosteroids (budesonide, prednisone) are used as induction agents and are prescribed for short periods to address disease flares in both mild to moderate and moderate to severe patients. Corticosteroids are generally administered through an oral or rectal route of administration. Common side effects are mild to moderate in intensity and include UC, headache, nausea, mood changes and sleep changes. |
|
• |
Immunosuppressants (methotrexate, thiopurines) are used to wean patients off steroid use and rarely as independent maintenance drugs in moderate to severe patients. Immunosuppressants are orally administered but are systemically distributed agents. Common side effects include a decrease in the number of white blood cells (leucopenia), headache, rash, nausea, and dyspepsia (indigestion), alopecia (hair loss), mild increase in levels of liver enzyme (aspartate aminotransferase), peritoneal abscesses, abnormally low levels of the protein albumin in the blood (hypoalbuminemia). |
Moderate to severe UC
The following treatments are typically used in the treatment of moderate to severe UC:
|
• |
Biologics (anti-TNF, anti-IL-12/IL-23, anti-integrin) are the primary maintenance therapy in moderate to severe UC and comprised 59% in 2016 of the UC therapeutic market. Biologics are injectable therapies and can be divided into two classes: those targeting cytokines, comprising 80% of the biologics market in UC, and those targeting cell trafficking, comprising the remaining 20%. Side effects include leucopenia, immunosuppression, cancer, infection and death. |
|
• |
JAK inhibitors (tofacitinib) are an induction and maintenance therapy for severe patients that do not respond to other therapies, including biologics. Similar to immunosuppressants, approved JAK inhibitors are oral agents that are systemically distributed. Common side effects of JAK inhibitors include headache and nasopharyngitis, immunosuppression, increased risk for infections, changes in serum lipid levels, mild neutropenia and anemia, and slight increase in the incidence of malignancies (lymphoma, breast cancer, and lung cancer). |
16

We believe that current therapeutics for the treatment of both mild to moderate and moderate to severe UC have the following limitations that we believe BT-11, if approved, may address:
|
• |
Safety and tolerability concerns. The majority of approved therapeutics for maintenance of UC (biologics and JAK inhibitors) are systemically distributed, resulting in effects on the immune system outside of the gastrointestinal tract. These effects result in increased risks for cancers, infections, blood clots and death. Given the chronic nature of these indications, there is a need for a safer long-term option. Despite a lower severity of disease, many of the mechanisms of action for current drugs in mild-to-moderate space are also tied to known toxicities such as drops in white blood cell counts and increased risk for infection in the case of immunosuppressants, and bone loss, weight gain, lowered quality of life and cardiovascular complications in the case of extended corticosteroid use. Thus, it is difficult to develop drugs with better safety profiles without targeting innovative mechanisms. |
|
• |
Inconvenient and costly route of administration. The main class of therapy in UC is biologics, which are injectable therapies administered through either intravenous or subcutaneous routes. Often, this requires a patient to visit a clinic or specialist care provider which disrupts daily life, increases health care costs and lacks convenience. Aminosalicylates (5-ASAs), the recommended therapy for mild-to-moderate UC by the American Gastroenterological Association, requires 2.4 to 4.8 grams per day, amounting to two to six tablets daily when dosed orally. Depending on the response to oral dosing, rectal dosing of mesalamine is sometimes required. |
|
• |
Limited efficacy. Resulting from a lack of innovation that repeatedly aims at very narrow targets, current therapeutics in UC have continually faced a capped effect size, achieving no greater than 25% in remission rate. For biologics, in placebo-controlled pivotal studies, the 4-domain remission rate effect size for biologics ranged between approximately 10% and 25%. To illustrate, the Gemini 1 trial for Entyvio provided a remission rate of 16.9% and the Gemini 1 and 2 trials for Humira provided 18.5% and 16.5% remission rates, respectively. Additionally, among patients responsive to treatment with biologics, approximately 40% will lose response within three years due to immunogenicity, development of compensatory inflammatory signaling, or unknown causes. Marketed and late-stage drugs in JAK and sphingosine-1-phoshate, or S1P, classes have achieved similar rates in Phase 3 studies including tofacitinib (18.5% remission in the Octave I trial) and ozanimod (18.4% in a trial conducted by the True North Center). Studies of biomarker normalization in recent studies support these rates. For fecal calprotectin, various cut-off values have been used to predict clinical remission including 250 µg/g such as for ustekinumab (30.3% normalization rate, 8.5% placebo adjusted at week 8) and 150 µg/g such as for vedolizumab (29.3% normalization rate, 12.5% placebo adjusted at week six). Similarly in a real-world scenario, tofacitinib had a 29% normalization rate using a 250 µg/g cutoff. In a complex autoimmune disease with multiple aspects to its pathogenesis, targeting a singular inflammatory pathway may continue to be ineffective in providing extended remission to more than 25% of patients. |
17
|
• |
Therapeutic gap. Currently no FDA-approved drugs address the region of the treatment paradigm between the aminosalicylate (5-ASA) failures and biologics, where the majority of patients exist. Patients are faced with the option of staying on a sub-optimal therapy, to which they are losing response, or moving into a class of treatment with drastically higher safety risks. Despite early promise, the emergence of safety concerns for JAK inhibitors and S1P modulators have limited the non-biologic immunomodulators that are currently in development to a biologic-failure class of patients. There is an unmet need for a safer and effective drug that can bridge the treatment paradigm from 5-ASA to biologics. |
Our solution for treatment of UC
We believe that BT-11, our lead oral UC product candidate, if approved, has the potential to treat patients prior to their progression into biologics and address the main limitations of current therapeutics based on the clinical and preclinical data to date. We believe BT-11, if approved, may offer the following advantages:
|
• |
Gut-restricted PK with low systemic exposure. In preclinical and clinical studies, a comparison between the gastrointestinal PK of BT-11 versus the systemic PK in five species (mice, rats, dogs, pigs and humans) indicated that 99% of orally administered BT-11 remains in the gut. In our Phase 1 clinical trial, we validated the translation of the gut-restricted PK in humans since gastrointestinal concentrations of BT-11 human participants were observed to be on average 6,000-fold higher in the colon than the maximum serum concentration, or Cmax, in blood. In these studies, we observed that BT-11 was highly stable in the gastrointestinal environment and reached the colon in high local concentrations that scale proportionally with oral dose. |
|
• |
Tolerability. In its Phase 1 clinical trial, BT-11 had no differences in presentation of adverse events, or AEs, clinical chemistry, changes in white blood cell counts, electrocardiogram, or ECG, and other clinical measures at doses up to 100 mg/kg (approximately 7,500 mg). Chronic studies in rats for six months and in dogs for nine months have been completed with NOAEL greater than or equal to 1,000 mg/kg, a dose which is 70-fold higher than the highest dose tested in our Phase 2 clinical trials in UC. |
|
• |
Convenient, once a day oral dosing. We designed BT-11 for IBD to be a single, once-a-day oral tablet. In our ongoing Phase 2 clinical trial in patients with mild to moderate UC, we are evaluating BT-11 at two doses (500 and 1,000 mg), each of which are contained in a single tablet. |
|
• |
Preclinical and translational testing. We have evaluated BT-11 in five mouse models of colitis and a pig model of colitis, and have observed consistent reduction of inflammatory markers and improvement of histological scores across diverse methods of disease induction including genetic, immunological, bacterial and chemical. In side-by-side comparative preclinical testing, BT-11 greatly exceeded the efficacy of other therapeutics (anti-TNF, 5-ASA, tofacitinib) in reduction of disease activity index, leukocytic infiltration and fecal calprotectin in Mdr1a-/- model of colitis. We believe this robust dataset lends substantial support to the validity of LANCL2 as an innovative immunometabolic pathway, with the potential to be targeted via BT-11 without inherent toxicities and which has demonstrated potent therapeutic activity in a broad range of animal models of IBD. |
18
|
• |
Innovative immunometabolic target not tied to toxicities. We have designed BT-11 to activate a novel target, LANCL2, to induce immunometabolic effects that favor regulatory responses. In human primary cells and preclinical disease models, BT-11 activated oxidative phosphorylation and promoted FOXP3 stability to enhance the suppressive activity of Tregs and restore local tissue homeostasis and immune tolerance. In addition to potentially being a very powerful monotherapy, given the novel target and lack of toxicities, we believe there is also potential to use BT-11, if approved, as a complementary or combination UC therapy. |

19
Phase 2 clinical trial of BT-11 in UC
Phase 2 clinical trial design
In August 2019, we initiated dosing in a Phase 2 randomized, placebo-controlled, double-blind, multicenter clinical trial of 195 patients with mild to moderate UC. The primary objective of this trial was to establish efficacy and safety of oral BT-11. The study’s primary endpoint measured clinical remission in mild to moderate UC patients at week 12 of BT-11 treatment initiation, as defined by a 3-component definition of rectal bleeding = 0, stool frequency = 0 or 1, and endoscopic subscore = 0 or 1. The Mayo score is the primary evaluation measure in UC clinical trials, serving as a composite score (0-12) of endoscopic appearance, stool frequency, rectal bleeding and physician assessment. The graphic below shows the design of the Phase 2 clinical trial.

A total of 195 patients with mild to moderate UC (total Mayo Score 4-10; Mayo endoscopic subscore [MES] ≥ 2) were targeted to be randomized in a 1:1:1 ratio to receive BT-11 low-dose (500 mg), BT-11 high-dose (1,000 mg) or placebo. In total, 198 patients were randomized with each of the treatment arms comprised of 66 patients. The randomization was stratified by whether the patient has had prior exposure to biologic therapy for UC and whether the patient had corticosteroid use at baseline. The trial consisted of a 28-day screening period, a 12-week induction period, an 18-week maintenance period and a two-week post-treatment safety follow-up period. Patients that were in clinical response or remission at week 12 were eligible to continue into the maintenance period. Patients entering the maintenance period stayed in the same blinded group assignment given in the induction phase.
The trial included endoscopies at baseline, week 12 (end of induction) and week 30 (end of maintenance). Endoscopic scoring was centrally read. During each endoscopy, biopsies were collected for histology, PK and target engagement in addition to other mechanistic and translational evaluations. Blood and stool samples were collected throughout the trial (baseline, weeks two, six, 12, 18, 24 and 30) to assess biomarkers, including calprotectin levels in feces. Rectal bleeding and stool frequency sub-scores were recorded by patient diary.
Secondary endpoints include endoscopic remission rate at week 12 (as defined by a MES of 0 or 1), mucosal healing rate at 12 weeks (defined by an MES of 0 or 1 and a Geboes Histologic Index score of less than 3.1), mean change in fecal calprotectin from baseline over 12 weeks, concentration of BT-11 in feces over 12 weeks and number of participants with treatment-related AEs.
20
Phase 2 clinical trial topline results
At week 12, in the intent-to-treat population, BT-11 induced clinical remission in 31.8% of patients at 1,000 mg QD and 30.3% at 500 mg QD, compared to 22.7% at placebo. BT-11 had a well-tolerated safety profile with no meaningful difference in AEs relative to placebo during the induction period.
|
|
|
|
Primary endpoint definition |
Clinical remission at Week 12 as defined by stool frequency of 0 or 1, rectal bleeding of 0 and Mayo endoscopic subscore of 0 or 1 |
|
|
|
|
Analysis population |
All randomized subjects |
|
|
|
|
Analysis method |
Stratified Cochran-Mantel-Haenszel Method |
|
|
|
|
Planned stratifications |
Previous biologic usage |
|
|
|
|
|
|
|
Placebo |
BT-11 500 mg |
BT-11 1000 mg |
|
Clinical remission (%) |
22.7 |
30.3 |
31.8 |
|
P Value |
— |
0.340 |
0.235 |
In the intent-to-treat population, we observed a positive trend in absolute clinical remission rates as defined by the 3-component modified Mayo Score, using a stool frequency subscore of 0 or 1, a rectal bleeding subscore of 0 and an endoscopic subscore of 0 or 1 following 12 weeks of oral treatment with BT-11 at 1,000 mg QD or 500 mg QD compared to placebo (31.8% and 30.3% versus 22.7%; p=0.340 and 0.235). The resulting placebo-adjusted clinical remission rates of 9.1% and 7.6% for the 1000 and 500 mg dose groups, respectively, and are consistent with standard of care treatments in both mild-to-moderate and moderate-to-severe UC. In a more moderate subset of patients (with Mayo score equal to or greater than 7 at baseline) the placebo-adjusted clinical remission rates were 11.5%; (p=0.153) and 8.7% (p=0.273) for the 1000 (n=47) and 500 mg (n=44) dose groups, respectively, as compared to placebo (n=50). Additionally, in a small subset of biologic experienced patients, positive placebo-adjusted remission trends were also observed (66% and 33% in the 1,000 (n=3) and 500 mg (n=3) cohorts, respectively, as compared to placebo (n=3, 0%)).
|
Clinical Remission
|
Clinical Remission
|
Clinical Remission
|



The conventional method for measuring the statistical significance of a result is known as the “p-value,” which represents the probability that random chance caused the result (e.g., a p-value = 0.001 means that there is a 0.1% or less probability that the difference between the control group and the treatment group is purely due to random chance). Generally, a p-value less than 0.05 is considered statistically significant, and may be supportive of a finding of efficacy by regulatory authorities.
21
During endoscopy at baseline and week 12, biopsies were collected for histological assessment of the colon. Histological samples were scored by a blinded central reader by the Geboes score, which is a histological scoring system for UC. The cutoff for histological remission was set as a Geboes score less than 2B.1, which is indicative of a lack of erosion, ulceration or granulation of the tissue, no crypt destruction, an absence of neutrophils in the epithelium and no noticeable increase of neutrophils in the lamina propria. In subjects with increased chronic inflammatory infiltrate at baseline, histological remission occurred in 42.1% of patients treated with 500 mg BT-11 (Δ = 16.6; p = 0.069) and 38.5% of patients treated with 1,000 mg BT-11 (Δ = 13.0; p = 0.158). In all randomized subjects, histological remission occurred in 48.5% of patients treated with 500 mg BT-11 (Δ = 13.7; p = 0.112) and 43.9% of patients treated with 1,000 mg BT-11 (Δ = 9.1; p = 0.285). Additionally, in a small subset of biologic experienced patients, positive placebo-adjusted histological remission trends were also observed (33% in both the 1,000 mg (n=3) and 500 mg (n=3) cohorts, respectively, as compared to placebo (n=3, 0%)).

Pharmacokinetic measurements confirmed that dose proportional increases of BT-11 were observed in stool samples across each dosing group. As illustrated below, oral dosing of BT-11 resulted in stable stool concentrations of approximately 1,600 µg/g at a 500 mg oral dose and 2,600 µg/g at a 1,000 mg oral dose over the 12-week induction period. Similar dose proportionality occurred in biopsy samples collected at week 12, resulting in 3.84 µg/g with the 500 mg oral dose and 5.91 µg/g with the 1,000 mg oral dose. Estimated maximal plasma concentrations were not different than those observed in normal healthy volunteers and displayed no dose proportionality, suggesting no evidence of greater systemic absorption with a compromised intestinal epithelial layer. No change in estimated maximal plasma concentrations occurred through the 12-week induction phase.
|
Stool Concentration
|
Colon (Tissue) Concentration
|
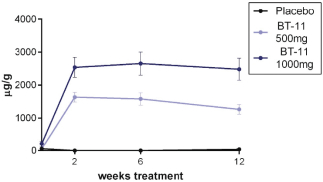

In the overall modified intent-to-treat population, which included all randomized patients in the trial, BT-11 treated patients were more likely to experience normalization of fecal calprotectin at each time point than patients receiving placebo, providing a response as early as two weeks post-treatment, as illustrated below. No significant differences in baseline fecal calprotectin were observed across groups (BT-11 high dose: 403 ± 382 g/g; BT-11 low dose: 462 ± 468 µg/g; placebo 543 ± 420 µ/g [mean ± SD]).
22
Fecal Calprotectin Normalization

As illustrated below, in patients with elevated fecal calprotectin levels (≥ 250 µg/g) at baseline, normalization and mean change in fecal calprotectin was assessed. Normalization of fecal calprotectin levels, commonly cited to be one of the most predictive biomarkers of response to treatment in UC and CD, was detectable in a subset of patients (n=106) with abnormal baseline calprotectin levels (>250 ug/g), after 2 weeks of oral dosing in over 40% of patients treated with BT-11 (n=64), at either dose level, when compared to 21% of patients receiving placebo (n=42). More specifically, normalization of fecal calprotectin occurred in 43.8% (Δ = 22.4) of patients receiving BT-11 1,000 mg and 40.6% (D = 19.2) of patients receiving BT-11 500 mg after 2 weeks of treatment, compared to 21.4% of patients receiving placebo. On average, patients receiving BT-11 1,000 mg experienced a mean change of -270.0 µg/g (Δ = -160.6) and patients receiving BT-11 500 mg experienced a mean change of -254.9 µg/g (Δ = -145.5), in each case after two weeks of treatment, compared to patients receiving placebo, who experienced a mean change of -109.4 µg/g. No significant differences in baseline fecal calprotectin were observed across the treatment groups in the subgroup analysis (BT-11 high dose: 461 ± 397 µg/g; BT-11 low dose: 613 ± 471 µg/g; placebo 639 ± 422 µg/g [mean ± SD]). Notably, in patients with elevated fecal calprotectin at baseline, BT-11 induced a 13.1 placebo-adjusted clinical remission rate at week 12.
|
Fecal Calprotectin Normalization
|
Fecal Calprotectin
|


23
In biopsies sampled from the most afflicted area of the colon, BT-11 increased the proportion of CD25+ regulatory CD4+ T cells (Treg) relative to baseline. BT-11 also provided a significant reduction in TNF+ myeloid cells relative to baseline when compared to placebo.

Among the analyzed populations by flow cytometry, an increase in IL10+ anti-inflammatory cells was correlated to induction of clinical remission in BT-11 treated groups. No association was observed in placebo, suggesting a BT-11 dependent effect. In BT-11 treated groups, macrophages (CD64+ CX3CR1+) were observed to be key producer of IL10. The increase in IL10-producing macrophages in response to BT-11 treatment was significantly (p = 0.0378) associated to lower disease activity scores. Interestingly, similar increases of IL10-producing macrophages were observed in mouse models of IBD in response to oral BT-11 treatment.
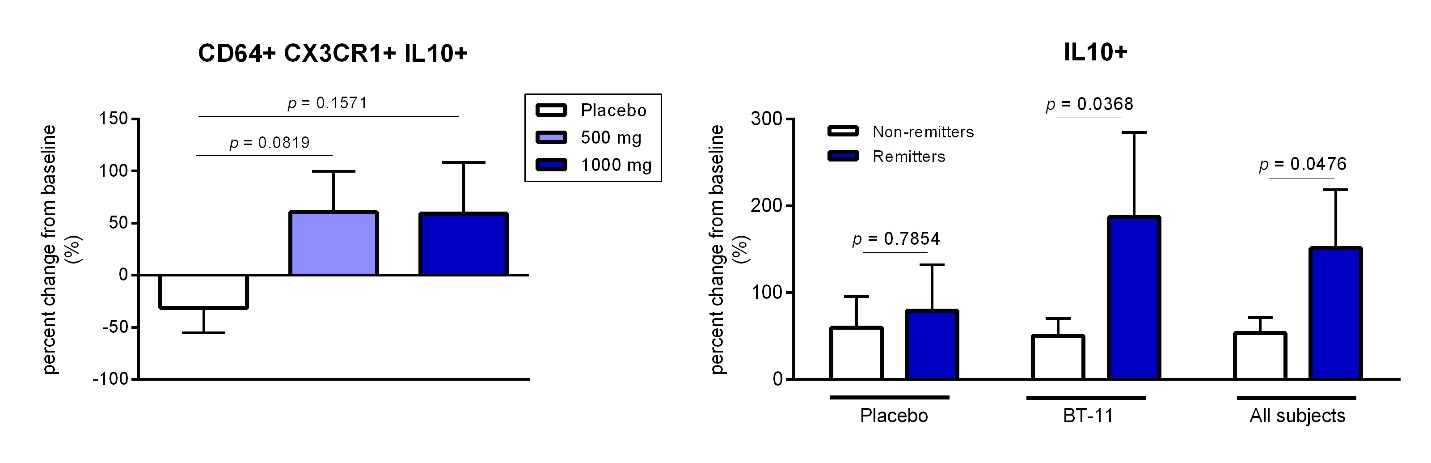
The AEs observed were similar across the three arms of the trial, with 30.3% of the placebo group, 27.3% of the 500 mg BT-11 group, and 30.3% of the 1,000 mg BT-11 group reporting one or more AEs. The most common AE observed was worsening of ulcerative colitis, reported in 9.6% of patients study-wide, with no meaningful differences between the two treatment groups and the placebo group. No other AE was reported in more than 5% of patients. In total, 37 AEs were judged to be possibly or probably related to BT-11, with 11 of these events in patients treated with the highest dose of BT-11 (1,000 mg). No meaningful difference was observed in the presentation of infections and infestations between patients receiving any dose of BT-11 and placebo, with no patient dosed with BT-11 presenting with lymphopenia. Four total SAEs were reported, all of which were judged to be unrelated to BT-11. Overall, no emergent safety or tolerability concerns were identified.
Phase 1 clinical trial of BT-11 by single ascending dose and multiple ascending dose in normal healthy participants
The safety and tolerability of BT-11 was assessed in a two-stage, single-center, double-blinded, randomized, placebo-controlled clinical trial in healthy male and female volunteers. The single ascending dose stage consisted of five groups of eight healthy male and female participants per cohort, each receiving a single oral dose of BT-11 or placebo in a six-hour fasted state. The multiple ascending dose stage consisted of three cohorts of ten healthy male and female participants, each receiving an oral dose of BT-11 or placebo once daily for seven days. Doses were tested ranging from 500 mg to approximately 7,500 mg in the highest cohort of each stage.
24
Safety and tolerability were assessed through types and frequency of AEs, physical examination, ECGs, biochemistry, hematology and urinalysis. No trends in AE presentation were observed in comparison between BT-11 and placebo or between specific cohorts. No serious adverse events were reported. No AEs were identified to have a probable or definite relationship to the study drug. None of the clinical laboratory results, including serum chemistry, hematology, coagulation and urinalysis, outside the reference range were considered to be clinically significant in either the single ascending dose or multiple ascending dose stages. No changes from baseline in white blood cell count or percentages of specific immune cell subsets were observed in blood on day seven of the multiple ascending dose stage relative to placebo in any one cohort or across all participants who received BT-11. No changes in ECG parameters were assessed as clinically significant and no treatment-emergent AEs, or TEAEs, arose from abnormal ECG measurements.
|
Single Ascending Dose |
Multiple Ascending Dose |
||
|
|
|
|
|
|
White Cell Count |
C-Reactive Protein |
White Cell Count |
C-Reactive Protein |
|
|
|
|
|
|
|
|
|
|




Change from baseline in white blood cell counts and C-reactive protein levels during single ascending dose (n = 10, placebo; n = 6, active) and multiple ascending dose (n = 6, placebo; n = 8, active) arms of the Phase 1 study of BT-11 in normal healthy volunteers.
PK was assessed in blood, at multiple time points post-dosing, and in stool, on day seven in the multiple ascending dose stage. BT-11 concentrations were quantified within feces on day seven of the multiple ascending dose stage. Concentrations of 2.39 mg/g were observed in participants who received 500 mg of BT-11 daily for seven days. Stool BT-11 levels were observed to scale proportionally in a manner similar to the scaling of oral dosage. In contrast to stool concentrations, a plasma Cmax of 372 ng/mL at 1.19 hours post-dose was observed in participants receiving 500 mg daily for seven days, a value that is more than 6,000-fold lower than concentrations observed in feces. Plasma concentrations scaled at a less than dose proportional scale when comparing high-dose to low-dose, with only an 8.2-fold increase in AUC0-24 relative to a dosage increase of 14.3-fold. Plasma BT-11 was quickly cleared with half-life of 2.7 hours. In parallel with PK analysis, calprotectin concentrations were measured in feces. This study was not powered for detecting statistical differences in biomarkers. Mean fecal calprotectin was observed to be lower in the BT-11 500 mg cohort than those observed in placebo.
25
Mean fecal calprotectin (µg/g)
|
|
|
|
|
|
Placebo |
BT-11 (500 mg) |
|
Single ascending dose |
33.8 |
12.3 |
|
Multiple ascending dose |
34.8 |
6.4 |
|
Fecal BT-11 SAD, day 2 |
Fecal BT-11 MAD, day 7 |
|
|
|
|
|
|


Fecal concentrations of BT-11 on day 2 of single ascending dose (n = 6) and day 7 of multiple ascending dose (n = 8) arms of the Phase 1 study of BT-11 in normal healthy volunteers.
Clinical development plan
Phase 3 clinical trial design of BT-11 in UC
We intend to initiate an integrated Phase 3 clinical program of BT-11 in patients with mild to moderate UC in the second half of 2021, subject to review of the complete Phase 2 data and subject to FDA feedback in June of 2021. The Phase 3 program is expected to be two independent induction trials feeding into a single maintenance trial. All aspects of these trials will be randomized, placebo-controlled, double-blind, parallel-group multicenter designs. The purpose of these trials will be to evaluate the efficacy and safety of oral BT-11 in the induction and maintenance of remission in patients with mild to moderate UC. The trial will use a co-primary endpoint of clinical remission, as defined by a Mayo stool frequency subscore of 0, Mayo rectal bleeding subscore of 0 and a Mayo endoscopy subscore of 0 or 1, at week 12 and week 52. Secondary endpoints will be included to assess mucosal healing by endoscopic and histologic evaluation, improvement in the endoscopic appearance of the mucosa, maintenance of corticosteroid-free remission, clinical response, fecal calprotectin and changes in individual Mayo subscores.
An estimated total of 632 patients with mild to moderate UC (total Mayo Score 4-10; Mayo endoscopic subscore MES ≥ 2) and a 6-point Mayo score (sum of rectal bleeding and stool frequency) equal or greater than 2 will be randomized in a 1:1 ratio to receive BT-11 low-dose (500 mg) or placebo into the first induction trial. The second induction trial will include 720 patients (randomized 1:1) with matching treatment groups and inclusion and exclusion criteria. The protocol for the second trial will hold the analysis of the maintenance endpoints. The final number of patients may be adjusted based on the final data package from our Phase 2 clinical trial. The randomization will be stratified by prior exposure to biologic therapy for UC and corticosteroid use at baseline. We will implement a controlled stratification to ensure a minimum of 20% of the overall population and a maximum of 40% have previously received biologic therapy. The trial will consist of a 28-day screening period, a 12-week induction period, a 40-week maintenance period and a 2-week post-treatment safety follow-up period. A mandatory steroid taper will commence at week 12.
The trial will consist of endoscopies at baseline, week 12 (end of induction) and week 52 (end of maintenance). Endoscopic scoring will be conducted centrally. During each endoscopy, biopsies will be collected for histology, PK and target engagement in addition to other mechanistic and translational evaluations. Blood and stool samples will be collected throughout the study (baseline, weeks two, six, 12, 18, 24 and 30) to assess biomarkers, including fecal calprotectin. Rectal bleeding and stool frequency subscores will be recorded by patient diary.
The graphic below depicts the expected design of the Phase 3 clinical trial.
26
Expected Phase 3 trial design in UC

Preclinical results
BT-11 has been studied in five mouse models and one pig model of IBD. These five mouse models encompass diverse methods of disease induction including chemical (DSS), cellular (adoptive transfer), genetic (Mdr1a-/-, IL10-/-) and bacterial (Citrobacter rodentium). Key findings from our preclinical studies are summarized in the tables below.
Summary of disease activity and histopathology results from BT-11 in five mouse models of IBD
|
|
|
|
|
|
|
|
Model |
Challenge |
Dose |
Length of |
Reduction in |
Reduction in |
|
DSS |
DSS in drinking water |
8 mg/kg, oral |
7 days |
70%* |
50%* |
|
Adoptive transfer |
4x105 CD45RBhi CD4+ T cells by IP injection |
8 mg/kg, oral |
6 weeks |
65%* |
80%* |
|
Adoptive transfer |
4x105 CD45RBhi CD4+ T cells and 1x105 Treg by IP injection |
8 mg/kg, oral |
6 weeks |
88%* |
95%* |
|
Mdr1a-/- |
Spontaneous/genetic |
8 mg/kg, oral |
6 weeks |
73%* |
95%* |
|
IL10-/- |
Spontaneous/genetic |
8 mg/kg, oral |
12 weeks |
75%* |
40%* |
|
C. rodentium |
1x109 CFU/mouse (strain DBS100) |
8 mg/kg, oral |
3 weeks |
85%* |
50%* |
|
* |
P ≤ 0.05 |
Summary of changes in CD4+ T cells and neutrophils in colon from BT-11 in five mouse models of IBD, measured by flow cytometry
|
|
|
|
|
|
|
Model |
Reduction in Th1 |
Reduction in Th17 |
Change in Treg cells |
Reduction in neutrophils |
|
DSS |
47%* |
66%* |
71%* |
42%* |
|
Adoptive |
22%* |
27%* |
29%* |
49%* |
|
Adoptive transfer (CD45RBhi, Treg co-transfer) |
46%* |
44%* |
68%* |
88%* |
|
Mdr1a-/- |
40%* |
35%* |
56%* |
79%* |
|
IL10-/- |
40%* |
35%* |
84%* |
61%* |
|
C. rodentium |
71%* |
87%* |
140%* |
95%* |
|
* |
P ≤ 0.05 |
27
Summary of colonic gene expression results from BT-11 in five mouse models of IBD
|
|
|
|
|
|||
|
Model |
Reduction in Tnf |
Reduction in Ifng |
Increase in Il10 |
|||
|
DSS |
76 |
%* |
47 |
%* |
118 |
%* |
|
Adoptive transfer |
19 |
% |
10 |
% |
88 |
%* |
|
Adoptive transfer (CD45RBhi, Treg co-transfer) |
31 |
%* |
69 |
%* |
145 |
%* |
|
Mdr1a-/- |
45 |
%* |
88 |
%* |
21 |
%* |
|
IL10-/- |
34 |
%* |
52 |
%* |
NA |
|
|
C. rodentium |
82 |
%* |
73 |
%* |
34 |
%* |
|
* |
P ≤ 0.05 |
In the DSS model, correlation between the colonic concentrations of BT-11 with key markers (fecal calprotectin, histologic score and TNF expression) of preclinical efficacy was tested at two doses levels (8 mg/kg, 25 mg/kg). In each marker, an inverse trend was observed with decreasing concentrations or scores with increasing BT-11 concentrations. Fecal calprotectin had the strongest correlation to BT-11 concentration of the parameters tested. Overall, a dose dependent decrease was observed in the colonic expression of TNFα, IL6, and MCP1.
|
|
||
|
|
|
|
|
Colon Tnfa |
Colon II6 |
Colon Mcp1 |
|
|
|
|
|
|
|
|




Preclinical biomarker responses to oral BT-11 (8, 25 mg/kg) during DSS colitis (n = 10, * P ☐ 0.05).
In the Mdr1a-/- genetic model, BT-11 (oral, 8 mg/kg) was compared to anti-TNF (2 mg/kg, intravenous), 5-ASA (oral, 25 mg/kg), and tofacitinib (oral, 30 mg/kg). Treatments were administered for six weeks. Disease activity index, a composite measure on a scale of 0-4 of rectal inflammation, stool consistency, weight loss and overall physical condition, was monitored throughout the treatment period. BT-11 provided significantly greater response relative to vehicle and the three comparative therapies, based on a side-by-side comparison. After six weeks of treatment, colon sections were prepared for histology and graded for leukocytic infiltration. Stool was collected for measurement of calprotectin. In both measures, the scores of BT-11 were significantly lower than vehicle and the three comparative therapies, as shown in the graphs below.
28

Comparative efficacy in disease activity index, leukocytic infiltration, and fecal calprotectin with BT-11 treatment (8 mg/kg) relative to anti-TNF (2 mg/kg), 5-ASA (25 mg/kg) and tofacitinib (30 mg/kg) (n = 9, * P ≤ 0.05; ** P ≤ 0.01).
Early validation of the local GI PK was conducted in rodents. While informative, the gastrointestinal tract of rodents is significantly different from humans in transit time, gut microenvironment and overall gastrointestinal physiology. We took an additional step to validate the gut-restricted properties of BT-11 in pigs. Pigs have a nearly 1:1 human equivalent dosing ratio to humans. Twenty-four hours after dosing, colon tissue and colonic stool were collected and assayed for BT-11 content. We observed high, dose-dependent levels of BT-11 in both tissue and stool. The ratio of BT-11 between tissue and stool in pigs was observed to be 1:10. Peak concentrations of less than 150 ng/mL, 3,000-fold lower than tissue and 30,000-fold lower than stool concentrations, were observed in plasma one hour post-dosing. Minimal differences were observed between pigs with IBD and healthy pigs in absorption or local GI PK.

Plasma concentrations of BT-11 from 0 to 24 hours after oral dosing with immediate release tablets (n = 4). Colonic content (stool), ileum and colonic tissue concentrations of BT-11 24 hours after oral dosing with immediate release tablets (50, 100, 200 mg) (n = 4).
29
Plasma PK parameters of BT-11 in pigs
|
|
|
|
|
|
Parameter |
50 mg |
100 mg |
200 mg |
|
Cmax (ng/mL) |
64 |
54 |
94 |
|
t1/2 (hr) |
0.9 |
0.9 |
0.5 |
|
AUC (hr * ng/mL) |
245 |
200 |
287 |
|
Vd (L) |
0.29 |
0.35 |
0.54 |
In addition to the PK studies, we also used the pig IBD model to measure BT-11 efficacy. We used a seven-day DSS challenge with four groups: placebo and BT-11 at 50 mg, 100 mg or 200 mg, given orally in tablets. Colonic tissue and stool samples were collected at the end of the seven-day study. By histology, the amount of inflammatory cells in the colon was significantly decreased at 100 mg and 200 mg of BT-11. Calprotectin was reduced by approximately half in all BT-11-treated groups. Regulatory CD4+ T Foxp3+ cells that help control inflammation were increased in the colonic wall of all three BT-11 treated groups. As minimal increased in efficacy measures were observed beyond 100 mg, the 500 mg and 1,000 mg dose levels were chosen for preliminary examination in clinical studies based on the pigs used in this study being approximately one-tenth the size of an average human.

Reduction of leukocytic infiltration and fecal calprotectin with increase in colonic CD4+ FOXP3+ cells in pigs challenged with DSS for 7 days and treated with BT-11 tablets (50, 100, 200 mg) daily (n = 9, * P ≤ 0.05).
In initial translational studies, BT-11 was tested in vitro with lamina propria mononuclear cells, or LPMCs, isolated from colonic samples of UC donors. BT-11 significantly reduced TNFα+, IL-4+ and IFNγ+, at concentrations of 0.01, 0.1 and 0.5 µM. BT-11 also significantly increased FOXP3+ LPMCs at these concentrations, suggesting that BT-11 remained effective in cells isolated from the afflicted target area. While numerous factors differ between in vivo and in vitro environments, these experiments also provide preliminary insight into the extracellular concentrations of BT-11 necessary to induce immunological changes in human cells.
LPMCs were obtained from colonic resections of UC patients. Cells were treated for 24 hours ex vivo with BT-11. Flow cytometry was used to identify TNF+, IL4+, FOXP3+ and IFNg+ CD45+ cells. Statistical significance by treatment group (n = 8) is shown in the tables below.

Reduction of TNF+, IL4+ and IFNg+, and increase in FOXP3+ cells in LPMC of UC patients treated with BT-11 ex vivo (0.01, 0.1, 0.5 µM) (n = 6, * P ≤ 0.05). Results are expressed as % within CD45+ (hematopoietic immune cells) obtained from the colon.
30
Preclinical GLP toxicology results
In non-clinical repeat dose GLP toxicity studies, BT-11 showed no signs of toxicity at all tested doses. These studies have included three-month studies in rats and dogs, six-month studies in rats and nine-month studies in dogs, providing the necessary non-clinical data to support long-term dosing in humans. In these pivotal toxicology studies in rats and dogs, the NOAEL was greater than 1,000 mg/kg. In targeted studies for cardiovascular, respiratory and central nervous system effects, a NOAEL ≥ 1,000 mg/kg was also observed with no effects on ECG parameters, heart rate, respiratory rate, or functional observational battery. No target organ systems have been identified pre-clinically. BT-11 has not been identified to have any dose-limiting toxicities in animals.
The genotoxicity of BT-11 has been tested in one in vivo test and two in vitro assays, one of which utilized human cells. The genotoxicity studies have been the standard Ames, chromosomal aberration and micronucleus tests. None of these tests suggested mutagenic potential up to tested limit doses. We plan to conduct carcinogenicity studies of BT-11 in parallel with our planned Phase 3 clinical trial in UC.
BT-11 had minimal inhibitory effects on CYP enzymes and common drug transporters. Combined with limited overall systemic exposure and a high plasma protein, BT-11 has shown low potential for negative drug-drug interactions in our preclinical studies. BT-11 has a long metabolic half-life, but a short systemic half-life of less than three hours in PK studies. Analysis of urine and feces from toxicology studies in rats and dogs indicate that BT-11 is excreted unmetabolized through both routes.
Background on Crohn’s disease and current treatments
CD is a chronic, autoimmune, inflammatory bowel disease that causes inflammation, irritation, and ulcers in any segment of the gastrointestinal tract, but most commonly affects the end of the small bowel and the beginning of the colon. It can affect the entire thickness of the bowel wall, and inflammation of the intestine can skip, or leave normal areas in between patches of diseased intestine. Symptoms include abdominal pain, increased abdominal sounds, rectal pain and bleeding, bloody stools, diarrhea, fever, weight loss, and malnutrition. Having CD puts a patient at increased risk of developing colon cancer. CD is estimated to affect over 700,000 patients in the United States. The global therapeutic market for CD was approximately $9.6 billion in 2016, which is expected to grow at a compound annual growth rate of 3.7% between 2016 and 2026. Of this global market, the United States market comprised approximately $7.8 billion in 2016. The main current treatments for CD depend on the severity of the disease and are divided into four classes:
Mild to moderate CD
Treatment for mild to moderate CD includes:
|
• |
Aminosalicylates (5-ASAs) (mesalamine, sulfasalazine) can be prescribed to mild CD patients with colonic disease. Aminosalicylates are generally administered through an oral or rectal route of administration. Unlike UC, for which 5-ASA is commonly accepted as a first-line therapy, the efficacy of oral aminosalicylates in CD is controversial and has limited supporting evidence. Aminosalicylates are generally well-tolerated by patients in the treatment of IBD. Common side effects include headache, nausea, dyspepsia, flatulence and diarrhea. Rare but more serious side effects include pleuritis, pericarditis, myocarditis, pancreatitis, cholestic hepatitis, nephritis and renal dysfunction. |
|
• |
Corticosteroids (budesonide, prednisone, cortisone) are used as induction agents and are prescribed for short periods to address disease flares across the spectrum of severity in CD patients. Corticosteroids are administered through oral, intravenous and rectal routes of administration in the treatment of CD. Over one-third of CD patients are steroid-dependent defined as a relapse after dose reduction or within 30 days of stopping a steroid regimen. Common side effects are mild to moderate in intensity and include UC, headache, nausea, mood changes and sleep changes. |
|
• |
Immunosuppressants (methotrexate, thiopurines) are used as independent maintenance therapy in CD and can be used in combination with biologics. Immunosuppressants can be orally administered or injected and are systemic agents. Because of the AE profile, use of immunosuppressants can require frequent monitoring. Common side effects include leucopenia (a decrease in the number of white blood cells), headache, rash, nausea, and dyspepsia (indigestion), alopecia (hair loss), aspartate aminotransferase (mild increase in levels of liver enzyme), peritoneal abscesses and hypoalbuminemia (abnormally low levels of the protein albumin in the blood). |
31
Moderate to severe CD
Treatment for moderate to severe CD primarily consists of biologics.
|
• |
Biologics (anti-TNF, anti-IL-12/IL-23, anti-integrin) are the primary maintenance therapy in moderate to severe CD and comprised 59% in 2016 of the UC therapeutic market. Anti-TNF and anti-IL-12/IL-23 comprise 80% to 90% of the biologics used in CD. Anti-integrin biologics have a limited penetrance into the CD market. Biologics are injectable therapies. Biologics, particularly those targeting cytokines, have been associated with increased risks of cancer, infection and death. |
We believe that current therapeutic options have the following limitations:
|
• |
Safety concerns. The primary therapies in CD pose significant health risks. Anti-TNF and anti-IL-12/IL-23 biologics have been associated with higher risks for malignancies and infections. One of two anti-integrin therapies for CD has been associated with greater risk for progressive multifocal leukoencephalopathy. Immunosuppressants are associated with drop in white blood cell counts, liver damage and risk for infection. In addition to the common side effects of corticosteroid outlined above, extended corticosteroid use has been shown to contribute to bone loss, weight gain, lowered quality of life and cardiovascular complications. There are limited options available for patients and clinicians who prioritize safety and low risk for AEs when selecting therapies. |
|
• |
Limited efficacy. Current therapeutics have limited efficacy, with 18% to 25% effect sizes in induction of remission. For instance, up to 20% of CD patients are resistant to steroid treatment and have limited options for induction therapy. |
|
• |
Lack of long-term maintenance options. In placebo-controlled pivotal clinical trials, the most successful biologics achieve an approximate 18% to 25% effect size in Crohn’s Disease Activity Index, or CDAI, based remission. Additionally, half of patients who receive biologics as maintenance therapy experience a relapse of disease within one year. Immunosuppressive therapies have a greater risk of side effects that typically result in attempts to withdraw the therapy. Approximately one-third of patients experience a relapse shortly after withdrawal. These frequent disease flares result in greater steroid use which has been associated with a higher risk for developing refractory disease and for need of surgical resection. |
|
• |
Inconvenience. Current marketed biologics require injections, either through subcutaneous or intravenous routes, that can necessitate trips to clinics or care sites for administration, be disruptive to the lives of patients, and result in slight decreases in compliance. Current oral drugs for CD require high doses taken multiple times daily. |
|
• |
Therapeutic gaps. Gaps exist on both ends of the CD treatment paradigm. There is no consensus first-line therapy in CD. The main competition in biologic naïve patients are immunosuppressants. No current therapeutic effectively treats patients who are refractory to biologics. These patients require frequent steroids and surgery. |
Our solution for treatment of CD
We believe that BT-11, if approved, has the potential to serve as a first-line CD therapy, offering treatment for patients prior to their progression into biologics by addressing the main limitations of current therapeutics. We believe that an oral, gut-restricted small molecule delivered once daily in a singular tablet could serve as a first-line therapy in the moderate to severe CD treatment paradigm. In clinical trials to date, BT-11 has been observed to have the following characteristics in the treatment of CD:
|
• |
Gut-restricted PK with low systemic exposure. GI concentrations of BT-11 are on average 6,000-fold higher in the colon than in blood. A gut-restricted PK profile may assist in limiting the number of observed systemic adverse side effects. |
|
• |
Tolerability. Clinical studies (>7-fold) and preclinical studies (>100-fold) of BT-11 to date have not identified any dose-limiting toxicities well above the proposed therapeutic window. No systemic suppression of white blood cells, a common feature of biologics, immunosuppressant and other drugs in development, has been observed with BT-11. |
|
• |
Convenient, oral, once a day dosing. We have designed BT-11 to be a single, once-a-day oral tablet. We are evaluating BT-11 in our ongoing Phase 2 clinical trial in patients with CD at a 1,000 mg dose contained in a single tablet per day. |
32
|
• |
Preclinical and translational testing. BT-11 has been evaluated in five mouse models of IBD and a pig model of IBD providing consistent efficacy across diverse methods of disease induction including genetic, immunological, bacterial and chemical. Comparative efficacy studies in animal models indicate a doubling of response relative to anti-TNF. In human primary cells, BT-11 significantly downregulated TNF and IFNg at low concentrations. We believe that these preclinical results support advancement of BT-11 in the treatment of CD. |
|
• |
Innovative target. BT-11 is the first therapeutic candidate to target and activate LANCL2, a novel target, to induce immunometabolic effects. With the high frequency of switching treatments observed in IBD due to lack or loss of response, we believe additional differentiated mechanisms of action are needed. |
Phase 1 clinical trial results
The Phase 1 clinical trial of BT-11 in normal healthy volunteers tested oral doses up to 7,500 mg for up to seven days. No treatment-related adverse event differences were noted between all BT-11 cohorts and placebo.
Clinical development plan
Phase 2 clinical trial design of BT-11 in Crohn’s disease
Following our receipt of topline results from the ongoing Phase 2 clinical trial of BT-11 in UC, we plan to initiate a Phase 2 randomized, placebo-controlled, double-blind, multicenter clinical trial to evaluate the efficacy of oral BT-11 in moderate to severe CD. The primary objective of this trial will be to establish the efficacy of oral BT-11 in inducing clinical remission at week 12 in patients with moderate to severe CD. The co-primary endpoints will be proportion of patients achieving clinical remission at week 12 defined by CDAI of less than 150 and proportion of patients with an endoscopic response at week 12 defined by 50% reduction from baseline in the simplified endoscopic index of severity for Crohn’s disease, or SES-CD, score. The graphic below depicts the planned design of the Phase 2 clinical trial.
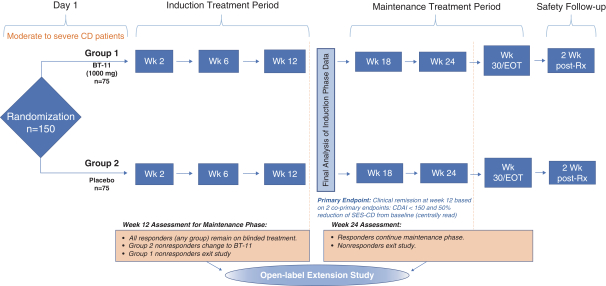
A total of 150 patients with moderate to severe, active CD will be randomized at a 1:1 ratio, in a centralized manner, to receive 1,000 mg of BT-11 or placebo. Moderate to severe, active CD will be defined as:
|
a) |
a CDAI score of 220 to 450; |
|
b) |
a patient-reported outcome (PRO-2) liquid/very soft stool frequency (SF) score ³ 4 and/or abdominal pain (AP) ³ 2, (seven-day average); and |
|
c) |
a SES-CD score ≥ 6 ( ≥ 4 for isolated ileitis) (confirmed by a central reader). |
33
Each of the treatment arms will comprise 75 patients. The randomization will be stratified by prior exposure to biologic therapy for CD with exposed population limited to 50% and corticosteroid use at baseline. The trial will consist of a 28-day screening period, a 12-week induction period, an 18-week maintenance extension period and a 2-week post-treatment safety follow-up period. Patients that are in clinical response or remission at week 12 will be eligible to continue into the maintenance period. Patients entering the maintenance period will stay in the same blinded group assignment given in the induction phase.
Secondary endpoints will include clinical response, endoscopic remission, deep remission, mean change in CDAI and mucosal healing. Biopsies collected from endoscopies conducted at baseline, week 12 and week 30 will be used to evaluate histology, PK, target engagement and other mechanistic and translational outcomes. Blood and stool will be used to assess biomarkers throughout the study.
Preclinical results
Many of our preclinical models of BT-11 evaluated the product candidate in both CD and UC, as described above. The adoptive transfer and IL10-/- models both develop ileitis in addition to colitis and would be considered the most relevant out of the five models of CD.
In human lamina propria mononuclear cells isolated from the colon of CD patients, BT-11 significantly reduced TNFα+ and IFNγ+ cells at concentrations of 0.1 and 0.5 µM. BT-11 significantly increased IL10+ cells at the same concentrations and increases FOXP3+ cells at 0.01, 0.1 and 0.5 µM.

Reduction of TNF+ and IFNg+, and increase in FOXP3+ and IL10+ cells in LPMC of CD patients treated ex vivo with BT-11 (0.01, 0.1, 0.5 µM) (n = 6, P ≤ 0.05). The results are expressed as % within CD45+ (hematopoietic immune cells) obtained from the colon.
Preclinical GLP toxicology results
Preclinically, pivotal repeat-dose studies in rats up to six months and dogs up to nine months have resulted in NOAEL ³ 1,000 mg/kg and no identified dose-limiting toxicities to date.
BT-11 for the treatment of eosinophilic esophagitis
Overview
BT-11 is an oral small molecule that is the first product candidate to target LANCL2 in the esophagus for EoE. In parallel to ongoing development in UC and CD, we are developing a modified formulation of BT-11 to treat EoE. EoE is categorized by inflammation of the esophagus due to an increased presence of eosinophils, a type of immune cell. The FDA has not approved any treatments of EoE. In addition, the FDA has granted orphan drug designation for product candidates designed to treat EoE, indicating that the FDA considers EoE to be an orphan disease. We have successfully filed an IND for BT-11 for the treatment of EoE in March of 2021.
Background on EoE and current treatments
EoE results from a dysregulation of Th2-mediated immunity that drives recruitment of eosinophils to the esophagus. The inflamed esophagus can cause chronic pain, frequent hospitalization and emergency room visits, difficulty eating and swallowing, and formation of fibrotic strictures. With up to 160,000 patients in the United States, EoE is an orphan disease without a currently FDA-approved therapeutic.
Currently, management of EoE often consists of the use of proton pump inhibitors, corticosteroids or inhaled medications typically used for asthma. It is estimated that approximately one-third of EoE patients have no response to current medical practices in EoE. Therapeutics in development are primarily corticosteroids, including budesonide which is used for IBD and asthma, and biologics, including dupilumab, which targets the IL-4 receptor for the treatment of asthma and eczema.
34
Our expectation is corticosteroid use and biologics will have similar limitations to those observed in IBD, including a presence of side effects and loss of response over time. Given the prevalence of EoE within both pediatric and adult populations, there is an unmet need for therapeutics with limited risk for side effects.
Our solution for the treatment of EoE
EoE stems from an excessive Th2 response in the esophagus that is thought to result from increased stimulation by epithelial cells and dendritic cells. In combination, regulatory functions of eosinophils, mediated through FOXP3, are suppressed in EoE patients. As such, there are multiple means to intercept the pathogenesis of disease.
Mechanistically, LANCL2 activation has the potential to impact the maintenance of the epithelial barrier, prevention of Th2 responses and improvement of suppressive capacity. We believe data generated from the evaluation of BT-11 efficacy in preclinical models of IBD and translational patient samples support that BT-11 may be used for the treatment of EoE. In mice, BT-11 was observed to decrease Th2 cells in the colonic mucosa, a mechanism important for both UC and EoE. In human LPMCs, BT-11 decreased the percentage of IL-4+ CD4+ T cells. IL-4 is one of the main cytokines produced by Th2 cells to recruit eosinophils.
As described previously, we believe that BT-11, if approved, has the potential to offer a differentiated profile to corticosteroids and biologics centering on tolerability, limited systemic exposure, oral dosing, and an innovative mechanism of action to restore tolerance.
Preclinical GLP toxicology
Oral BT-11 is supported by a robust GLP toxicology package potentially enabling chronic dosing through six-month rat and nine-month dog studies up to 1,000 mg/kg. No dose-limiting toxicities have been observed in safety pharmacology studies and no mutagenic potential has been suggested by genotoxicity studies.
Clinical development plan
We have successfully filed an IND for BT-11 and received FDA clearance for the treatment of EoE in March of 2021. This EoE trial will assess a modified formulation of BT-11-provided drug release in the oral cavity and esophagus. The primary objective of the trial will be to assess biomarkers of type 2 immune responses, analyze changes in esophageal eosinophil count and determine effects on the dysphagia symptom questionnaire, the primary patient-reported outcome measure in EoE. A secondary objective will be to assess PK of the modified formulation of BT-11 in humans.
BT-11 for the treatment of psoriasis and atopic dermatitis
Overview
BT-11 is a topical small molecule that is the first product candidate to target LANCL2 in the skin for psoriasis and atopic dermatitis. In parallel to ongoing development in intestinal disorders, we are developing a topical formulation of BT-11 for the treatment of skin inflammation. Psoriasis and atopic dermatitis are both disorders of the skin that cause itchiness, rashes, red patches, thickened skin and the formation of plaques or scales. We intend to submit an IND for BT-11 for the treatment of psoriasis in the second half of 2021.
Background on psoriasis and atopic dermatitis and current treatments
Psoriasis and atopic dermatitis are commonly linked to factors associated with both keratinocytes and immune cells resulting in inflammation of the skin with the formation of rashes and persistent itch. Psoriasis commonly manifests as plaque psoriasis in which the skin thickens and takes on a scaly appearance due to over-proliferation of skin cells. This over-proliferation results from an increase activation and reactivity of Th17 cells. Similarly, in atopic dermatitis, over-active Th17 cells strongly contribute to the proliferation of skin cells. The activation of Th2-related responses are also often tied to the immunological changes during active disease in atopic dermatitis. Psoriasis was estimated to afflict 14.9 million patients, resulting in a global market of $12.2 billion in 2017 and projected to be about $19.2 billion by 2027. Atopic dermatitis was estimated to afflict 81.6 million patients, with over a three-fold greater prevalence in children and adolescents, resulting in a global market of $6.4 billion in 2017.
35
Currently, management of psoriasis often consists of the use of topical corticosteroids or immunosuppressants. For more severe cases or those presenting with psoriatic arthritis, use of systemic immunosuppressants (methotrexate, cyclosporine) or a biologic targeting TNF or the IL-12/-23 pathway. In Phase 3 trials, ustekinumab had a 63.1% placebo adjusted response rate and adalimumab had a 64% placebo adjusted response rate, wherein the response was a 75% reduction in the psoriasis area and severity index. Atopic dermatitis has a similar treatment paradigm with the use of topical corticosteroids and calcineurin inhibitors as first-line drugs, followed by use of systemic immunosuppressants (methotrexate, cyclosporine) and biologics (dupilumab) targeting the IL-4 receptor. In pivotal trials, dupilumab had a 28% placebo-adjusted response rate, wherein the response was a score of 0 or 1 (clear or almost clear) on the Investigator’s Global Assessment and a reduction of 2 points or more in that score from baseline.
Similar to their usage in other autoimmune disease, biologics and immunosuppressants require monitoring of liver functions and immunosuppression, as use of these agents has been linked to increased risks of infections and cancers.
Our solution for the treatment of psoriasis and atopic dermatitis
We believe topical BT-11, if approved, could function as an alternative front-line therapy to reduce the usage of corticosteroids and immunosuppressants. In addition to no dose limiting toxicities identified in completed nonclinical and clinical trials, we believe that BT-11 through its Treg-associated mechanism of action could offer a greater likelihood of maintained responses relative to other topical therapies. We believe further applications of topical BT-11 as an adjunctive therapy to systemic treatments in refractory cases could be possible.
In an imiquimod-induced mouse model of psoriasis, BT-11 significantly decreased the presence of TNF+, IL-17+, IL-6+, and IL-21+ cells in the spleen when administered directly to the skin daily. Importantly, TNF and IL-17 are critical elements of the pathogenesis of disease in both psoriasis and atopic dermatitis.

Immune cell changes induced by topical BT-11 in an imiquimod-induced model of psoriasis after one week of treatment (n = 9, * P☐0.05).
Preclinical GLP toxicology
Oral BT-11 is supported by a robust GLP toxicology package potentially enabling chronic dosing through six-month rat and nine-month dog studies up to 1,000 mg/kg. No dose-limiting toxicities have been observed in safety pharmacology studies and no mutagenic potential has been suggested by genotoxicity studies. The oral and general toxicity package will be supported by in vitro and in vivo skin corrosion and irritation assays.
Clinical development plan
We intend to submit an IND for BT-11 for the treatment of psoriasis in the second half of 2021, and to initiate a Phase 1b clinical trial in psoriasis in the second half of 2021. This trial will assess a topical formulation of BT-11 to be applied directly to the skin. The primary objective of the trial will be to assess cytokine biomarkers of immune responses, analyze changes in epidermal thickness and determine effects on the psoriasis area and severity index score, the common primary outcome measure in psoriasis. A secondary objective will be to assess PK of the topical formulation of BT-11 in humans.
36
BT-104 for the treatment of lupus
Overview
BT-104 is an orally-active, systemically bioavailable small molecule therapeutic candidate that targets LANCL2 that we are developing for the treatment of SLE. The pathogenesis of SLE is connected to defective apoptosis leading to stimulation of B cells by dendritic cells and CD4+ T cells to produce auto-antibodies. These antibodies activate the complement system and deposit in organs leading to inflammation and tissue damage. We believe the activation of LANCL2 can intercept these events upstream through skewing of CD4+ T cells to regulatory phenotypes and maintenance of the metabolic requirements for autophagy. SLE remains a poorly-managed disease with safety concerns, limited efficacy and lack of diversity in current treatment options. We believe that an oral molecule with a differentiated mechanism of action could address the limitations of current therapies. We intend to submit an IND for BT-104 for the treatment of SLE in the second half of 2021.
Background on SLE and current treatments
SLE involves a complex cascade of events that results in auto-antibody generation and immune complex formation. The deposition of these complexes manifests local inflammation in the tissues most commonly associated with lupus, kidney, cardiovascular system and skin, leading to damage and diminished function over time. SLE is an expanding public health threat that affects over 1.5 million patients in the United States and 5 million people worldwide, according to the Lupus Foundation of America. Direct health care costs amount to $18 billion per year in the United States and up to $12 billion in lost productivity as a result of the disease. Sales of the top 10 therapeutics for lupus amount to over $4 billion annually.
Nearly 60% of SLE patients experience more than one flare per year or present with persistently active disease. Mortality rates and progression to end stage renal disease, or ESRD, have plateaued in recent years with currently available treatment options, indicating an unmet clinical need for safer and more effective therapeutics. SLE is commonly treated with corticosteroids, with 87% of patients being placed on a steroid-based regimen. Other therapeutic options fall into four classes:
|
• |
Antimalarials (hydroxychloroquine) have been used in the treatment of SLE due to an ability to lower the IFNγ response. Up to 70% of SLE patients are estimated to be on antimalarials. Due in part to approval in 1956 for SLE, robust knowledge on the efficacy of antimalarials in SLE through placebo-controlled clinical trials is limited in regards to currently relevant endpoints. |
|
• |
Biologics (belimumab, rituximab) used in the treatment of SLE primarily target B cells. Belimumab is the only new FDA-approved treatment for SLE in the past 60 years and inhibits BAFF to block B cell maturation. In Phase 3 testing, the high dose of belimumab had an effect size less than 10% relative to placebo in the primary endpoint of SLE responder index response rate. |
|
• |
Immunosuppressants (mycophenolate mofetil (MMF), methotrexate) are often used in the treatment of SLE and function by broadly blocking the proliferation of B and T cells. While used in a large portion of patients with lupus in combination with other therapies, the main clinical trials for immunosuppressants in lupus have failed to demonstrate effects in approvable endpoints by the FDA. |
|
• |
NSAIDs are used to manage fever, arthritis and headaches coupled with SLE. Due to an association with renal side effects, the use of NSAIDs in the treatment of SLE is closely monitored. Cutaneous reactions and hepatotoxicity are also observed with higher frequency in SLE patients. |
SLE remains a poorly managed disease with a lack of diversity in therapeutic options. Aside from limited efficacy, current medications are required to be taken multiple times daily in high doses and complex combinations. Limitations of current therapeutics include:
|
• |
Safety concerns. The use of corticosteroids for the treatment of SLE is associated with numerous side effects including cataracts, osteoporosis, and cardiovascular damage. With common doses of steroids ranging from 7.5 to 100 mg, it has been noted that a 1 mg/day increase in steroid exposure results in a 2.8% increase in risk for new organ damage in SLE. In clinical trials of belimumab, more death was observed in the treated groups relative to placebo, including due to septic shock, pneumonia and suicide. Antimalarials and immunosuppressants both increase risk of cardiovascular complications and infections. |
37
|
• |
Disease progression. In as little as 10 years post-diagnosis, SLE patients accrue neuropsychiatric, renal, cardiovascular and musculoskeletal damage that cause chronic pain in two-thirds of patients. This damage accrual leads to lower quality of life and increased health care burden. Development of lupus nephritis occurs in 50% to 70% of patients and ESRD, necessitating kidney transplants, occurs in 15% to 30% of patients due to poor disease management with current therapeutics. |
|
• |
Lack of new therapeutic options. Only one new drug for SLE has been approved in the last 60 years, with many clinicians instead choosing the off-label use of sub-optimal drugs. Current mechanisms of action target broad immunosuppression, interferon production, activation of B cells or the production of antibodies. With all current therapies targeting similar pathways, it is likely that subpopulations of SLE are continually non-responsive even with switches in medication. |
Our solution for treatment of SLE
The common investigational therapeutic targets in SLE are B cells and plasma cells, interferon responses and production of antibodies, which ignore the cascade of upstream events that lead to their activation. LANCL2 has the potential to intercept the complex cascade of upstream events that result in auto antibody generation and immune complex formation at two levels. First, as previously described in the context of BT-11, LANCL2 is an important receptor in regulatory CD4+ T cells and is differentially expressed by over 7-fold in Tregs relative to non-Treg CD4+ T cells. Activation of LANCL2 boosts oxidative phosphorylation and synergizes with IL-2/CD25 signaling to support and enhance FOXP3 stability and the suppressive function of Tregs. In human SLE and the NZB/W mouse model of lupus, CD25+ FOXP3+ CD127low Tregs are decreased in number and exist with a less suppressive phenotype. IL-2 levels are also suppressed in SLE, leading to CD25- Tregs that fail to enact regulatory functions. Preclinically in animal models, neutralization of CD25 has led to accelerated disease while the adoptive transfer of Tregs slows disease onset.
Second, LANCL2 supports the process of autophagy, the process that allows cells to eliminate defective cell material. In SLE, impaired autophagy results in the incomplete processing of apoptotic cells and accumulation of nuclear antigen that immune cells begin to react to over time. In SLE, impairment of LC3-associated phagocytosis proteins, such as Atg7 and Rubicon, results in lesser lysosomal fusion and the production of inflammatory cytokines during a process that typically produces IL-10. In the absence of LANCL2, macrophages and dendritic cells increase phagocytic uptake of cells but have lower expression of many autophagy-related genes. Critically, autophagy is a highly energy-intensive process, requiring mitochondrial metabolism to break down the material. The activation of LANCL2 can facilitate the generation of mitochondrial energy and enhance the production of IL-10 in phagocytes.
We believe that BT-104, if approved, may address the limitations of current therapeutics in the treatment of SLE based on the following characteristics below.
|
• |
Enhanced tolerability. There have been no dose-limiting toxicities to date in clinical development of BT-11. As a regulatory receptor, its activation does not result in the suppression of white blood cells or greater susceptibility to infection or malignancies. Preliminary dose-range finding studies with BT-104, at doses of 125 mg/kg, 250 mg/kg and 500 mg/kg, has resulted in no adverse effects in rats at up to seven days of dosing. |
|
• |
Halt of disease progression. No currently available therapy aims to restore homeostasis or address the continued presentation of nuclear antigens. Therefore, compensatory mechanisms may develop over time, resulting in loss of efficacy. The activation of LANCL2 may aid in reversing the original break in immune tolerance. Preclinically, LANCL2 activation has been observed in mice to prevent the worsening of proteinuria with interventional dosing of BT-104. |
|
• |
Differentiated mechanism of action. Current therapeutics for the treatment of SLE are all immunosuppressive in their mechanism of action, making LANCL2 clearly differentiated. LANCL2 induces regulatory CD4+ T cells and ameliorates metabolic dysfunction in immune cells. While differentiated, LANCL2 has reduced the production of anti-dsDNA antibodies and expression of IFNγ, in mouse models and human primary cells. |
38
Preclinical results
In a NZB/W F1 mouse model of lupus, BT-104 reduced serum anti-dsDNA antibodies and prevented worsening of proteinuria grade from baseline. Mice were treated with BT-104 daily for 12 weeks between the ages of 24 and 36 weeks. Ninety percent of mice treated with BT-104 experienced an improvement or no change in proteinuria grade from baseline, in comparison to 90% of vehicle treated controls that experienced a worsening in grade. Grade 2 or lower proteinuria is well correlated with the prevention of ESRD clinically.

Reduction of serum anti-dsDNA antibodies and prevention of proteinuria worsening after oral treatment of NZB/W F1 with BT-104 (20 mg/kg) for 12 weeks between the ages of 24 and 36 weeks (n = 10, P ☐ 0.05).
Oral BT-104 treatment has resulted in immunological changes systemically. In the spleen, 20 mg/kg of BT-104 increased CD25+ FOXP3+ Tregs and decreased IL21+ T follicular helper (Tfh) cells. Tfh cells assist in B cell maturation and amplify humoral responses.

Oral treatment of NZB/W F1 mice with BT-104 (20 mg/kg) for 12 weeks, between the ages of 24 and 36 weeks, increased the percentage of CD4+ Treg cells and suppressed T follicular helper cells in the spleen (n = 10, P ≤ 0.05).
The efficacy of LANCL2 activation was evaluated in cells obtained from blood (PBMCs) of SLE patients with active disease. BT-104 was observed to have a dose-dependent response, with maximum efficacy observed between 100 nM and 200 nM when PBMCs were treated with agents that activate inflammation, namely PMA + ionomycin (general activation), TLR7 (gardiquimod) and ODN2395 (type C CpG). BT-104 prevented IFNγ secretion in human primary cells stimulated with potent IFNγ inducers.
39
Ex vivo efficacy of BT-104 in human PBMCs
|
|
||||||
|
IFNγ, % reduction from vehicle |
||||||
|
Stimuli |
50 nM BT-104 |
100 nM BT-104 |
200 nM BT-104 |
|||
|
PMA (5 ng/mL) + Ionomycin (500 ng/mL) |
51 |
* |
54 |
* |
70 |
* |
|
Gardiquimod (2 µg/mL) |
42 |
* |
90 |
* |
92 |
* |
|
ODN2395 (2.5 µg/mL) |
33 |
|
46 |
* |
59 |
* |
|
* |
P ≤ 0.05 |
Preclinical GLP toxicology results
Preliminary dose-range finding studies have been conducted with oral BT-104 at doses of 125 mg/kg, 250 mg/kg and 500 mg/kg in rats over seven days. These studies revealed a NOAEL ³ 500 mg/kg with no changes in hematology, biochemistry, organ weights, macroscopic or microscopic lesions. PK studies of BT-104 in rats show a bioavailability greater than 70% within the therapeutic dose range of 1 mg/kg to 20 mg/kg.
In preparation for IND submission, BT-104 will be assessed in 28-day repeat dose toxicity studies in rats and dogs at doses of 250 mg/kg, 500 mg/kg and 1,000 mg/kg.
Clinical development plan
We intend to submit an IND for BT-104 for the treatment of SLE in the second half of 2021 and initiate a Phase 1 clinical trial in normal healthy volunteers in 2022. This study will feature a standard single ascending dose/multiple ascending dose design (five cohorts of single ascending dose and three cohorts of multiple ascending dose) testing a dose range 5-fold to 10-fold greater than the projected clinical therapeutic dose. The primary objective of the study will be to assess the safety and tolerability of BT-104 by frequency and severity of AEs, standard safety laboratory results, ECG and vital signs. A secondary objective will be to assess the PK of BT-104 in humans.
BT-104 for the treatment of rheumatoid arthritis
Overview
BT-104 is a systemically bioavailable small molecule product candidate that targets LANCL2 that we are developing for the treatment of rheumatoid arthritis. The pathogenesis of rheumatoid arthritis is linked to dysfunction of CD4+ T cells leading to over-activation of cells within the joint synovium. LANCL2 is an anti-inflammatory receptor that controls CD4+ T cell subsets towards regulatory phenotypes. The activation of LANCL2 results in a net decrease of inflammatory cell infiltration. Rheumatoid arthritis is a chronic autoimmune disease with therapeutic gaps resulting from safety concerns and limited efficacy of current treatment options. We believe that an oral molecule with a differentiated mechanism of action could address the limitations of current therapies for the treatment of rheumatoid arthritis. We intend to submit an IND for BT-104 the treatment of rheumatoid arthritis in the second half of 2021.
Background on rheumatoid arthritis and current treatments
Rheumatoid arthritis is an autoimmune disease that results in chronic pain and loss of mobility in joints due to excessive inflammation that swells the joints and erodes bone and cartilage. Rheumatoid arthritis affects 1.3 million patients in the United States and has a global market estimated to be approximately $25 billion annually. With age as a risk factor, the number of new cases of arthritis is expected to increase as the elderly population expands. The therapeutic market is projected to have a compound annual growth rate of over 4% from 2018 to 2025.
Rheumatoid arthritis requires chronic treatment to maintain remission and treatment escalation or add-ons to address flares. Approximately 10% of patients will be in remission for a full year. Current treatments for rheumatoid arthritis are categorized into five classes:
|
• |
NSAIDs (ibuprofen, naproxen) are used in mild cases of rheumatoid arthritis to provide slight anti-inflammatory effects in addition to pain and symptom relief. Safety risks for NSAIDs are minor in comparison to other classes of drugs but possess limited efficacy in moderate to severe cases. |
|
• |
Corticosteroids (prednisone) are used to broadly suppress inflammation in the presence of a flare. Corticosteroid use in the treatment of rheumatoid arthritis is associated with weight gain, muscle loss, decrease in bone density and cardiovascular issues. |
40
|
• |
Disease-modifying antirheumatic drugs (DMARDs) are a broad class of drugs including methotrexate, leflunomide, sulfasalazine, and hydroxychloroquine. |
|
• |
Biologics (anti-TNF, anti-CD20, anti-IL6R, fusion protein) for the treatment of rheumatoid arthritis are diverse with targets of cytokine signaling, B cell depletion, and Th17 differentiation. Biologics can result in increased risk of cancers and infections. |
|
• |
JAK inhibitors (tofacitinib, baricitinib) function by blocking the signals that promote activation of CD4+ T cells and other immune cells. The side effects of JAK inhibitors are numerous, including drop in white blood cell counts, anemia, blood clotting and death. |
The limitations of current therapies in rheumatoid arthritis are tied to safety and efficacy. There is a high risk of severe side effects with all current treatments, aside from NSAIDs. Medical practice lacks an option for moderate to severe patients that does not present with significant risk of co-morbidities. Given “black box” safety warnings on both biologics and JAK inhibitors, we believe new therapies that are safer and equally effective would have a clear market advantage. About 40% to 50% of rheumatoid arthritis patients experience a 50% drop in severity when switching to a new therapy with 30% to 35% entering remission. The high non-response rates and loss of response rates in rheumatoid arthritis continues to offer ample market opportunity for novel mechanisms of action.
Our solution for the treatment of rheumatoid arthritis
We believe BT-104, if approved, may offer a competitive advantage relative to current therapies given its novel mechanism of action and the lack of dose-limiting toxicities we have observed to date preclinically. LANCL2 is a key receptor in the plasticity between Treg and Th17 cells and accumulation of Th17 cells in the synovium is a shared characteristic of most individuals with rheumatoid arthritis, which drive the production of TNF and the recruitment of neutrophils and fibroblast like synoviocytes. Interactions between these three cells types are central to joint swelling, pannus formation and osteoclast differentiation.
Due to an ability to inhibit de novo Th17 differentiation and to prevent adoption of effector phenotype in Tregs, the activation of LANCL2 shifts the balance of this plasticity towards Tregs. Clinically, in the context of BT-104, LANCL2 activation has reduced calprotectin levels, a marker of neutrophil infiltration comprised of S100A8 and S100A9 subunits that are greatly increased in rheumatoid arthritis. Preclinically, LANCL2 activation significantly decreases calprotectin levels relative to tofacitinib, a marketed therapy for arthritis, in a separate model of autoimmune disease. These combined immunological changes can decrease ICAM1 and CD109 overexpression that leads to excessive fibroblast like synoviocyte infiltration.
In preliminary clinical development and preclinical studies, LANCL2 activation posed significantly less risk for toxicities than biologics, JAK inhibitors and other DMARDs. LANCL2 activation is novel in comparison to other treatments in rheumatoid arthritis by initiating an immunometabolic signaling cascade to enhance regulatory and tolerogenic pathways. This could offer an additional treatment option to the high refractory and relapsing population in rheumatoid arthritis.
Preclinical GLP toxicology results
BT-104 has been tested in a seven-day dose range finding study in rats up to doses of 500 mg/kg with no adverse effects observed. We expect to initiate GLP 28-day repeat dose toxicity studies in rats and dogs in the first half of 2021.
Clinical development plan
Subject to receiving authorization to proceed under an IND, we expect to initiate a safety and tolerability Phase 1 study in normal healthy volunteers of BT-104 in 2022.
BT-111 for the treatment of nonalcoholic steatohepatitis (NASH)
Overview
BT-111 is a small molecule product candidate that targets LANCL2 that we are developing for the treatment of NASH. NASH results from activation and proliferation of fibroblasts in response to chronic inflammation caused by obesity, lipid accumulation and insulin resistance. LANCL2 activation may ameliorate autoimmune disease and has shown benefit in restoring insulin sensitivity in obesity. We intend to submit an IND for BT-111 in the second half of 2021.
41
Background on NASH and current treatments
NASH affects over 8 million patients in the United States and greater than 16 million patients globally. With current NASH-related health care costs exceeding $15 billion in the United States, NASH represents a large public health problem. With expected yearly growth of nearly 45%, NASH may become one of the most pressing health concerns worldwide. NASH increases risk for liver failure, hepatocellular carcinoma and cardiac death.
There is no currently FDA-approved treatment for NASH. As such, the projected annual market opportunity by 2025 is estimated to be up to $30 billion. Current treatment options for NASH are diet and exercise, which have poor patient adherence in both the short- and long-term. Further, these approaches are more effective in non-alcoholic fatty liver disease, or NAFLD, whereas reversal of the fibrosis occurring in NASH may require pharmacological intervention.
A primary challenge in the development of NASH therapeutics has been the identification and selection of quantitative clinical trial endpoints and lack of information on representative disease-related biomarkers to use in early-stage clinical trials. Current therapeutics under development have effect sizes ranging between 8% and 15% relative to placebo fibrosis improvement with no worsening of NASH, suggesting sufficient room for improvement. We believe the ideal NASH therapeutic would be an oral medication capable of inducing immunological and metabolic effects.
Our solution for the treatment of NASH
NASH is a reversible fibrotic disorder of the liver resulting from chronic inflammation and metabolic dysfunction. Obesity and type 2 diabetes are major risk factors causing lipid accumulation and local macrophage activation that stimulates myofibroblasts to overproduce collagen and other extracellular matrix proteins. LANCL2, in addition to immune cells, is expressed within hepatocytes and muscle cells. In the context of type 2 diabetes, the natural ligand of LANCL2 improves glycemic control, increases glycogen metabolism and lessens adipose tissue inflammation.
Within the liver, the loss of LANCL2 increases liver size, triglyceride content and fibrosis. Mechanistically, LANCL2 activation increases expression of Pgc1a in metabolic tissues resulting in increased mitochondrial biogenesis and synergizes with insulin to activate glycogen synthase enzymes. Combined, these effects can reverse metabolic dysfunction and restore lipid homeostasis. The potential efficacy of LANCL2 in NASH is additionally supported by the anti-inflammatory effects, including downregulation of TNF and IL-6, that can reduce the proliferation and activation of myofibroblasts in the liver.
There are currently no FDA-approved therapies for the treatment of NASH. We believe BT-111, if approved, has the potential to address the unmet clinical need in NASH by providing:
|
• |
Well-tolerated with convenient dosing. The activation of LANCL2 has not been associated with any dose-limiting toxicities in preclinical or clinical development in NASH or other indications. LANCL2 activation does not block lymphocyte trafficking or proliferation directly, making it unlikely to be associated with drops in white blood cell count or increased risk for bacterial or viral infections. BT-111 would be administered as a once-daily oral tablet that is generally accepted to result in high patient compliance. |
|
• |
Dual impact on immunity and metabolism. The anti-inflammatory actions of LANCL2 are well established, with an ability to decrease TNF expression and differentiation of Th1 and Th17 cells in multiple disease models. Within adipose tissue, LANCL2 decreases the presence of TNF producing macrophages. The activation of LANCL2 also increases glucose uptake and utilization in muscle and maintains insulin sensitivity. BT-111 may therefore address the inflammation-associated fibrosis and the underlying metabolic dysfunction in NASH. |
Preclinical results
Therapeutic dosing of BT-111 (10 mg/kg) for six weeks between weeks six and 12 of CDAA diet, reduced lipid accumulation and liver fibrosis (n = 10, P ≤ 0.05). Liver fibrosis, assessed by percent positive area in liver histology by Masson’s trichrome staining, was approximately normalized to standard diet controls.
42
Efficacy of BT-111 in CDAA diet-induced model of NASH
|
|
|
|
|
|
Vehicle, percent change |
BT-111 (20 mg/kg), percent |
|
Liver triglycerides |
+81% |
+34%* |
|
Masson’s trichrome |
+106% |
+9%* |
|
* |
P ≤ 0.05 |
In the same model and treatment paradigm, BT-111 reduced markers of inflammation, including Th17 cells and TNF-producing myeloid cells (n = 10, P ≤ 0.05). Both IL-17 and TNF have been linked to the progression of NAFLD to NASH due to their ability to induce hepatic stellate cell proliferation and activation, even in the presence of low TGF-ß.

Reduction of Th17 and CD11b+ TNF+ cells after oral treatment of BT-111 (10 mg/kg) for 6 weeks in a 12 week CDAA-diet model of NASH (n = 10, P ≤ 0.05).
Preclinical GLP toxicology
We plan to test BT-111 in a seven-day dose range finding study in rats up to doses of 1,000 mg/kg in the fourth quarter of 2020. We expect to conduct PK studies testing the bioavailability and profile of BT-111 in rats in the therapeutic window (1-20 mg/kg) in the second half of 2020. We intend to initiate GLP 28-day repeat dose toxicity studies in rats and dogs in the second quarter of 2021.
Clinical development plan
Following an expected IND submission in the second half of 2021, we intend to initiate a Phase 1 study in normal healthy volunteers in 2022. This study will feature a standard single ascending dose/multiple ascending dose design testing a dose range 5-fold to 10-fold greater than projected therapeutic dose. The primary objective of the study will be to assess the safety and tolerability of BT-111 by frequency and severity of AEs, standard safety laboratory results, ECG and vital signs. A secondary objective will be to assess the PK of BT-111 in humans and metabolic biomarkers including cytokine and lipid profiles.
BT-111 for the treatment of type 1 diabetes
Overview
BT-111 is a small molecule product candidate that targets LANCL2 that we are developing for the treatment of type 1 diabetes. Type 1 diabetes is an autoimmune disease in which insulin-producing pancreatic beta cells are destroyed, requiring life-long insulin replacement therapy. No treatment exists to maintain or restore beta cell mass. LANCL2 is a receptor that induces anti-inflammatory and regulatory effects that may help to restore self-tolerance and that promotes glycemic control. We intend to submit an IND for BT-111 in the second half of 2021.
43
Background on type 1 diabetes and current treatments
Type 1 diabetes is a devastating autoimmune disease that results in the destruction of cells within the pancreas that produce insulin leading to elevated blood sugar and damage to other organs. Type 1 diabetes afflicts about 1.5 million patients in the United States and 3.3 million globally. Its incidence is estimated to increase by 3.9% annually worldwide between 2016 and 2026. Direct medical expenses related to the management of type 1 diabetes, including insulin, syringes, pumps and monitoring supplies, and annual lost income totaled $16 billion in the most recent estimate according to the Juvenile Diabetes Research Foundation. Total type 1 diabetes-related health care costs commonly exceed $100,000 per patient over his or her lifetime.
The current treatment of type 1 diabetes is management with insulin therapy. Use of combination therapies to improve glycemic control are rare. No treatment is currently available to prevent or delay progression of beta cell loss upon diagnosis. Due to the difficulty of managing glycemia and complications from the disease, the average patient lifespan is reduced by over 17 years. The limitations of standalone insulin therapy are:
|
• |
Variability. Highly individualized glycemic responses to meals, responsiveness to the type of insulin and variation in physical activity confound proper control over blood glucose management. Current approaches place a high responsibility on the patient to accurately estimate insulin need. |
|
• |
Failure to address all ß cell functions. Insulin therapy does not address additional missing functions of ß cells, such as the production of C-peptide or amylin, the auto-reactivity of the immune system, or the complications that arise. |
|
• |
Long-term complications. The current recommended HbA1c goal for type 1 diabetes patients is 7.0. However, the average HbA1c is 8.4 in type 1 diabetes. This deviation from the target range has severe long-term consequences and confirms the need for additional glycemic control measures to combine with insulin therapy. Prolonged dysregulated blood glucose can lead to obesity and insulin resistance with age, worsening management and contributing to the deterioration of health. |
Our solution for the treatment of type 1 diabetes
At diagnosis, type 1 diabetes patients typically still have between 20% and 40% of their original ß cell mass and function. Following the beginning of insulin therapy, endogenous insulin production is observed to be increased in a 12-month window post-diagnosis, suggesting the ability to regenerate or maintain pancreatic function. The combination of ß cell loss remission and the need for precipitating events suggest that the immunological destruction of pancreatic islets is capable of being stalled or even reversed.
We believe the activation of LANCL2 has a potential 3-fold benefit in type 1 diabetes through promotion of anti-inflammatory responses, maintenance of beta cell health and support of glycemic control. LANCL2 down-regulates the production of TNF, IFNy and IL-21, the main cytokines associated with islet destruction. In type 1 diabetes, ß cells undergo Bax and caspase 3 induced cell death. LANCL2 activation increases calcium signaling and reduces mitochondrial stress, reducing the activation and expression of Bax and caspase 3. LANCL2 assists in glucose uptake in muscle and other metabolic tissues, promoting translocation of GLUT4 and glycogen synthetase activity, which can aid in glucose homeostasis in situations of low insulin production.
Plasma concentration of the natural LANCL2 ligand, ABA, is decreased in diabetes, removing an endogenous compound that contributes to insulin sensitivity and glucose uptake. ABA can be produced in the central nervous system and pancreas, among other locations. Metabolic benefits of the LANCL2 signaling axis are observed in hepatocytes, adipocytes and myocytes through uptake and oxidation of glucose to improve the systemic glucose homeostasis. LANCL2 activation may alleviate pancreatic stress in early disease and assist with glucose control in late disease. We believe BT-111 may be used in type 1 diabetes to:
|
• |
Provide anti-inflammatory effects. Increased number of IL-10-producing cells during the short window post-diagnosis is associated with better blood glucose control. Similarly, type 1 diabetes patients with lower IFNγ production were more likely to exhibit an extended post-diagnosis remission in ß cell loss. LANCL2 activation has been shown to increase the ratio of IL-10 CD4+ T cells to IFNγ CD4+ T cells. |
|
• |
Preserve ß cell functions. BT-111 maintains ß cell mass in a NOD mouse model and reduces apoptosis of human islet cells in response to oxidative and inflammatory stress in vitro. |
44
|
• |
Normalize post-prandial spikes in blood glucose. Type 1 diabetes patients experience prolonged elevations in blood glucose after meals and overall non-physiological trends. The activation of LANCL2 may assist in insulin-independent glucose uptake and energy storage that would provide greater control of blood glucose regulation. |
Preclinical GLP toxicology
We intend to initiate GLP IND-enabling studies for BT-111 in 2021.
Clinical development plan
We intend to initiate a safety and tolerability study of BT-111 in normal healthy volunteers in 2022.
Our NLRX1 pathway product candidates
The NLRX1 pathway
The NLRX1 pathway addresses the immunometabolic interface from a different angle than the LANCL2 pathway. As a mitochondrial associated receptor, NLRX1 is favorably positioned to induce both metabolic and immunological effects. NLRX1 is unique among its family of receptors as one of three out of 23 NLRs to primarily induce regulatory and anti-inflammatory effects. The most commonly studied NLRs are those associated with the inflammasome, a complex whose activation results in high levels of cytokine production and cell death that increases inflammation. An overactive inflammasome and polymorphisms in inflammasome associated NLRs, such as NLRP1 and NLRP3, are commonly identified in autoimmune and chronic inflammatory diseases. NLRX1 is the natural counterbalance to this process, serving to control and negatively regulate many of the processes induced by inflammasome activation. NLRX1 serves as a key regulatory molecule permitting oxidative metabolism without the generation of inflammatory responses to the resultant by-products. Through effects on c-Abl and Nrf2, NLRX1 activates expression of enzymes to increase intracellular antioxidant capacity. The downregulation of intracellular reactive oxygen species and lactate production decreases NF-κB activity, a main signaling element upstream of many inflammatory cytokines. As a result, NLRX1 activation decreases a wide range of cytokines, including ones of CD4+ T cell origin as well as ones of myeloid origin.
|
•NX-13 multi-modal mechanism of action by activating the NLRX1 pathway •Decrease cellular reactive oxygen species •Antagonism of NF-κB activation resulting in downregulation of myeloid cell and T cell derived cytokines like TNF and IFNγ •Decreased inflammasome formation (NLRP1 and NLRP3) •Decreased differentiation of effector CD4+ T cells. •NLRX1 activation in intestinal epithelial cells increases mitochondrial metabolism and prevent oxidative stress. •Favors cell survival, the maintenance of barrier integrity and the expression of tight junction proteins. |
|

At the cellular level, NLRX1 activation results in shifts in the balance in CD4+ T cell subsets, decreased activation of macrophages and decreased stress-induced cell death for epithelial and other specialized cells. The decrease in lactate production and NF-κB activity results in proportionally higher Treg cells relative to Th17 cells with NLRX1 activation. Similarly, NF-κB is a main molecule that signals the polarization of macrophages into inflammatory subsets as opposed to those associated with tissue repair and homeostasis. In macrophages, NLRX1-associated ubiquitination of mitochondrial antiviral-signaling protein (MAVS) is thought to contribute to decreased activation. In intestinal epithelial cells, airway epithelial cells and neurons, NLRX1 activation increases mitochondrial metabolism and prevents oxidative stress. These effects are beneficial to functions of these cell types, including cell survival, the maintenance of barrier integrity and expression of tight junction proteins. In addition, the added metabolic support decreases the likelihood that a cell will exacerbate inflammation during cell death. By favoring apoptosis, a relatively silent form of cell death, NLRX1 can aid in the tissue homeostasis and repair process to prevent chronic tissue damage and fibrosis.
45
Our development pipeline of NLRX1-targeting product candidates has a preliminary focus on autoimmune and inflammatory diseases most closely associated with altered inflammasome activation. We are currently developing three product candidates targeting the NLRX1 pathway: NX-13, NX-66 and NX-73. Further, we believe the development of NLRX1 ligands for IBD, CNS disorders, including MS, and respiratory disorders including asthma is supported by specific preclinical and translational target validation.
NX-13, an oral NLRX1 agonist for the treatment of UC and CD
Overview
NX-13 is an orally active small molecule product candidate that targets NLRX1, a negative regulatory NOD-like receptor. NX-13 is the first product candidate to target NLRX1. We have completed a Phase 1 clinical trial for NX-13 in normal healthy volunteers.
Our solution for treatment of UC
Due to the involvement of microbial and dietary factors in the onset and progression of UC, many pattern-recognition receptors, which are part of the environmental surveillance system by detecting bacterial and food components, have been implicated in the pathogenesis of UC. Unlike many of its family members, NLRX1 has neither well-characterized and prominent genetic mutations nor inflammatory effects. Out of 23 human NLRs, three are categorized as regulatory NLRs, of which NLRX1 is one. While NLRX1 interacts with many of the same downstream signaling elements as other NLRs, NLRX1 serves to control and mitigate these responses rather than activate them. This includes control of reactive oxygen species (ROS) and NF-κB signaling, leading to a downstream decrease in TNF-α, IL-6, and IFN-g production and a lessening of mucosal inflammation. Mechanistically, the activation of NLRX1 could have a 3-fold benefit in UC with the ability to modulate epithelial integrity, host-microbiome interactions, and mucosal immune responses. As such, we believe NX-13 could provide significant benefits compared to current therapeutics for UC.
46
NX-13 is designed to target NLRX1 and provide therapeutic efficacy by inducing anti-inflammatory effects in CD4+ T cells and other immune cells of the gastrointestinal (GI) tract. NLRX1 was identified as an immunometabolic therapeutic target based on loss of function characterization in animal models of IBD in which the loss of NLRX1 increased disease severity and histological lesions, altered CD4+ T cell differentiation, disrupted epithelial barrier integrity, and influenced gut microbial populations. Through the immunometabolic actions of NLRX1 leading to increased oxidative phosphorylation and decreased NF-κB activity in CD4+ T helper (Th) cells, NX-13 inhibits the differentiation of Th1 and Th17 subsets and overall immune activation. NX-13 is a gut-restricted compound with minimal systemic absorption observed in preliminary pharmacokinetic studies in rats. This allows for oral activity in the gastrointestinal tract.

As a once-daily, oral, gut-restricted molecule, we believe NX-13 has a promising target product profile for the treatment of UC. Early nonclinical and clinical studies of NX-13 have not revealed any dose-limiting toxicities. In preclinical comparative efficacy studies to 5-ASA, anti-TNF, and tofacitinib, we have observed significantly greater changes in disease activity, biomarkers and histological parameters with NX-13. We believe that through NLRX1 activation, NX-13, if approved, may offer the potential for an independent or complementary approach to address the limited efficacy observed in UC with current options.
Phase 1a clinical trial of NX-13 by single ascending dose and multiple ascending dose in normal healthy participants
In July 2020, we initiated dosing in a Phase 1a clinical trial of NX-13 in normal healthy volunteers. This trial was a randomized, double-blind, placebo-controlled single and multiple ascending dose trial to evaluate the safety, tolerability and PK of oral NX-13. The primary objective of the study was to assess the safety and tolerability of single and seven-day repeat oral doses of NX-13. Safety and tolerability endpoints included changes from baseline in biochemistry, hematology, urinalysis, and ECG parameters, vital signs and frequency and severity of AEs. The secondary objective of the study was to assess the PK of NX-13 following administration of single and seven-day repeat oral doses. Plasma PK was obtained to assess systemic exposure. Stool concentrations of NX-13 were measured for local PK to inform doses carried forward into future UC patient studies. The graphic below depicts the design of the Phase 1 clinical trial.
47

The single ascending dose arm consisted of 35 healthy volunteers in a total of five cohorts (250 to 4,000 mg). Each cohort enrolled seven participants with five participants randomized to receive NX-13 and two participants randomized to receive placebo. The multiple ascending dose arm consisted of 21 healthy volunteers enrolled in a total of three cohorts (1,000 to 4,000 mg). Each cohort had seven participants with five participants randomized to receive NX-13 and two participants randomized to receive placebo. NX-13 was administered by oral tablet.
Across the eight cohorts, no SAEs were reported. No trends in laboratory tests were identified, including analysis of hematology, biochemistry, coagulation, and urinalysis parameters, at any tested dose. No meaningful changes in body weight, vital signs or ECG parameters were observed. The maximum tolerated dose was identified to be 10-fold greater than anticipated therapeutic dose.
Pharmacokinetic analysis identified that GI concentrations were more than 2,500-fold greater than peak plasma concentrations. As illustrated below, plasma NX-13 was observed to have a high distribution half-life with greater than an 80% reduction in peak plasma concentrations in three to four hours after dosing. After multiple dosing, no accumulation was observed and no change in trough concentrations were observed over the seven-day period. In plasma, peak concentrations were more than 2-fold less than dose proportional. In contrast, stool concentrations of NX-13 were observed to be dose proportional, as illustrated below.
|
Plasma NX-13
|
Stool NX-13
|


48
In addition to pharmacokinetics, stool samples were used to quantify fecal calprotectin levels, as illustrated below. Across all groups, 56% of subjects dosed with NX-13 were brought to or below the detection limit of the assay. In both mean concentrations and change from baseline, a dose response to NX-13 was observed.
|
Fecal Calprotectin
|
Fecal Calprotectin
|


Clinical development plan
In the first half of 2021, we plan to initiate a Phase 1b trial of NX-13 in active UC patients. The objective of the study will be to identify changes in endoscopic score, fecal calprotectin and patient reported outcomes after treatment with one of multiple dose levels of NX-13. The results of the Phase 1b trial will be used to appropriately power a Phase 2 trial, which we expect to initiate in 2022.

Preclinical results
NX-13 was evaluated in three mouse models of IBD to represent chemical, cellular and genetic methods of disease induction. NX-13 was administered orally at doses of 20 mg/kg in DSS and Mdr1a-/- and 10 mg/kg in adoptive transfer. Disease activity index, colonic leukocytic infiltration, lamina propria neutrophils and colonic TNF expression were used as endpoints.
Efficacy of NX-13 in three mouse models
|
|
|
|
|
|
|
Model |
Reduction in final |
Reduction in leukocytic |
Reduction in |
Reduction in Tnf |
|
DSS (n = 9) |
55%* |
76%* |
70%* |
59%* |
|
Adoptive transfer (n = 9) |
72%* |
52%* |
60%* |
79%* |
|
Mdr1a-/- (n = 9) |
68%* |
73%* |
60%* |
85%* |
|
* |
P ≤ 0.05 |
49
In the Mdr1a-/- model, concentrations of 24 cytokines were tested after six weeks of treatment with NX-13 at 10 or 20 mg/kg and compared to vehicle, NX-13 untreated, mice. Numerous inflammatory cytokines were observed to be significantly reduced including those tied to CD4+ T cell responses (IFNg, IL-17, TNFα, IL-4) as well as ones of myeloid origin (IL-6, IL-1ß, TNFα, MIP1α, MCP1).

Normalized concentrations of cytokines in the colon of Mdr1a-/- mice after six weeks of NX-13 treatment (0, 10, 20 mg/kg) (n = 9, * P ≤ 0.05 for high dose only, ** P ≤ 0.05 for low and high dose relative to vehicle).
The efficacy of NX-13 (10 mg/kg, oral) was compared to anti-TNF (2 mg/kg, iv), 5-ASA (oral, 25 mg/kg), and tofacitinib (oral, 30 mg/kg) in DSS and Mdr1a-/- mice. NX-13 treatment resulted in an 86% reduction in fecal calprotectin, a 64% reduction in Th1 cells, and a 64% reduction in leukocytic infiltration. The reductions observed with NX-13 were 1.5-fold to 2.5-fold greater than those observed with anti-TNF and tofacitinib.
Comparative efficacy of NX-13 to current IBD therapeutics
|
|
|
|
|
|
|
||||
|
Treatment |
Change in |
Change in lamina |
Change in leukocytic |
Treatment |
Change in |
||||
|
NX-13 |
-86 |
%* |
-64 |
%* |
-64 |
%* |
NX-13 |
-86 |
%* |
|
anti-TNF |
-32 |
%* |
-13 |
% |
-26 |
% |
anti-TNF |
-32 |
%* |
|
tofacitinib |
-26 |
%* |
-39 |
%* |
-45 |
%* |
tofacitinib |
-26 |
%* |
|
mesalamine |
+9 |
% |
-5 |
% |
-5 |
% |
mesalamine |
+9 |
% |
|
* |
P ≤ 0.05 |
50
We have tested the ability of NX-13 to reach the distal colon in high quantities after oral dosing in mice, rats and pigs. In pigs, which have a highly similar gastrointestinal tract to humans and an approximate 1:1 equivalent dosing ratio, NX-13 was observed at a concentration of over 6,000 ng/mg in colonic stool at 24 hours after oral dosing with 100 mg tablets. In our preclinical studies, one-seventieth of the colonic content NX-13 penetrated through the epithelial barrier. Dose-dependent concentrations are observed in both stool and tissue.

Colonic content (stool) and colonic tissue concentrations of NX-13 24 hours after oral dosing of pigs with immediate release tablets (10, 50, 100 mg) (n = 4).
The efficacy of NX-13 in pigs with DSS colitis was evaluated to colonic concentrations of NX-13 with efficacy markers. Oral NX-13 tablets protected against weight gain deficiency in a dose-dependent manner. The 100 mg dose of NX-13 resulted in a 74% reduction in leukocytic infiltration, a 54% reduction in fecal calprotectin and an 80% reduction in TNF expression relative to placebo. We also observed significant reductions in all three parameters at a 50 mg dose. Leukocytic infiltration and TNF expression were significantly reduced at a 10 mg dose. When compared to the local GI PK concentrations of NX-13, colonic concentrations greater than 45 ng/mg appear sufficient to induce response in all parameters. Additional magnitude of response can be achieved with concentrations greater than 85 ng/mg.

Reduction of weight gain deficiency, leukocytic infiltration, fecal calprotectin, and colonic TNF expression in pigs challenged with DSS for 7 days and treated with oral NX-
13 tablets (10, 50, 100 mg) daily (n = 6, P ≤ 0.05).
In PBMCs from moderate to severe UC patients (n = 6), NX-13 significantly reduces TNFα+ cells down with a 0.01 µM dose and IFNg+ cells down with a 0.05 µM dose.

Reduction of TNF+ and IFNγ+ in peripheral blood mononuclear cells from UC patients with ex vivo treatment of NX-13 (0.01, 0.05, 0.1 µM) after stimulation with PMA/ionomycin for 6 hours (n = 6, P ≤0.05).
51
Preclinical GLP toxicology results
NX-13 has completed IND-enabling studies and cleared 2 INDs plus initiated Phase 1 testing.
In rats, we evaluated doses of 250, 500, and 1,000 mg/kg/day in a 28-day GLP repeat-dose toxicity study. The NOAEL of NX-13 in rats was 1,000 mg/kg with no treatment-related AEs or clinical observations at any of the tested dose levels. No mortality or noteworthy findings were recorded. No differences in food consumption, ophthalmology, gross pathology, hematology, serum chemistry or histopathology were observed at the 1,000 mg/kg dose level compared to the control, and this dose level was the NOAEL.
In the 28-day repeat dose GLP toxicity study in dogs at 125, 250 and 500 mg/kg, no treatment-related ocular abnormalities, cardiovascular effects, clinical chemistry, hematology, coagulation, urinalysis, gross or microscopic findings were seen in either sex during the week 4 examinations and necropsy. No clinical signs were considered evidence of systemic toxicity. The NOAEL of NX-13 in dogs was 500 mg/kg.
Oral NX-13 displayed no signs of disruption of organ system function in in vivo evaluation of respiratory, cardiovascular and central nervous system function in GLP safety pharmacology studies up to tested limit doses. NX-13 did not show mutagenic potential in Ames, chromosomal aberration or micronucleus tests up to non-precipitating dose levels in vitro and 2,000 mg/kg in vivo.
Our solution for treatment of CD
NX-13 is an oral small molecule therapeutic targeting the NLRX1 pathway, a novel mechanism of action. The activation of NLRX1 is effective at inducing responses in CD4+ T cells, intestinal epithelial cells and myeloid cells. These responses are designed to result in a downregulation of multiple CD-associated cytokines (TNF, IL-1ß, Th1 cytokines), improved tolerance to the microbiome and reduced levels of oxidative stress. We believe NX-13, if approved, may address the unmet need for additional therapies in the treatment of CD based on the following observed characteristics:
|
• |
Well-tolerated, with no dose-limiting toxicities identified to-date. |
|
• |
Oral, once-daily tablet. |
|
• |
Greater preclinical efficacy than current therapeutics with robust validation in multiple models. |
In addition to the robust immunological changes induced by NLRX1 activation, NX-13 may also improve fibrosis, a key therapeutic gap in CD. Central to the fibrosis onset is the elevation of reactive oxygen species, which increases TGF-ß signaling, increases myofibroblast proliferation and induces epithelial-to-mesenchymal transition. NX-13 reduces ROS through an induction of antioxidant enzymes including glutathione peroxidase and thioredoxin reductase. The loss of NLRX1 has been shown to reduce markers of autophagy, including LC3-II and ATG16L1, in models of IBD. A SNP ATG16L1, well-correlated to an increased risk of CD, results in an active protein that is more susceptible to mitochondrial stress-induced degradation. Therefore, increased NLRX1 activity, which decreases mitochondrial and oxidative stress, could increase autophagy in CD by enabling stabilization of ATG16L1. Through this autophagy mechanism, NLRX1 further supports intestinal redox balance to promote tissue repair over fibrosis.
Clinical development plan
We have completed a Phase 1a clinical trial of NX-13 in normal healthy volunteers. We expect to initiate a Phase 2 proof-of-concept trial evaluating NX-13 in CD in the second half of 2022.
Preclinical results
Many of the preclinical models of CD are shared with that of UC. Data on three mouse models and a pig model of IBD are presented above. The adoptive transfer model develops ileitis in addition to colitis and would be considered to be the most relevant of the three models to CD.
Preclinical GLP toxicology results
Preclinically, pivotal repeat-dose studies in rats up to tested dose limits (1,000 mg/kg) for 28 days and dogs up to tested dose limits (500 mg/kg) for 28 days have resulted in no identified dose-limiting toxicities to date.
52
NX-66 for the treatment of multiple sclerosis
Overview
NX-66 is an oral, small molecule agonist of the NLRX1 pathway that we are developing for the treatment of MS. NLRX1 is a non-inflammasome-forming mitochondrial-associated NLR, expressed by immune cells systemically and in the CNS, that down-regulates inflammation in injury and autoimmune diseases, including models of experimental autoimmune encephalomyelitis, or EAE, colitis, and brain injury. Progressive MS presents with cortical lesions comprised of activated microglia and an overall increase in microglia in the brain. These microglia, as well as IL-12- and TNF-producing dendritic cells, contribute to direct neuronal damage as well as the ongoing demyelination that disrupts axonal architecture. In active EAE (direct immunization), passive EAE (adoptive transfer), and spontaneous EAE (2D2 mice), the loss of NLRX1 results in worsening of disease, greater microglial activation, and increased prevalence of spinal cord lesions. We intend to submit an IND for NX-66 in 2022.
Background on multiple sclerosis and current treatments
Multiple sclerosis is a chronic inflammatory, demyelinating and neurodegenerative disorder of the central nervous system. The initial diagnosis of multiple sclerosis is frequently characterized by episodes of neurological disturbances followed with residual deficits or full recovery (relapsing-remitting multiple sclerosis) and in a minority by a slow accumulation of disability from the onset (primary progressive multiple sclerosis).
Multiple sclerosis affects over 700,000 patients in the United States and over 2 million people worldwide. Multiple sclerosis results in decreased quality of life, with cognitive deficiencies reported in over 70% of patients and 42% of patients requiring informal caregiving from their families, according to the National Multiple Sclerosis Society. The global market for multiple sclerosis is currently over $20 billion per year and is expected to grow at a compound annual growth rate of 2.8% per year through 2022.
Relapsing-remitting multiple sclerosis is a fragmented market with all classes of therapies clustered around the similar efficacy profiles with 20% to 37% no evidence of disease activity rates, and patients with average age over 40 experience on average 25% or less inhibition of disability progression. In contrast, very few treatment options for progressive multiple sclerosis exist. The most recent approval for progressive multiple sclerosis, ocrelizumab, reported less than a 7% effect size relative to placebo in Phase 3 studies. Current therapeutics for MS are clustered into six main classes:
|
• |
Interferon betas were the first class of disease modifying therapeutics in multiple sclerosis and are believed to function through reduction of antigen presentation and CD4+ T cell proliferation. Clinical trials of interferon beta have shown no evidence of disease activity rate of approximately 20% of treated patients and have inhibited disability progression by 5% to 40%. |
|
• |
Biologics (alemtuzumab, natalizumab, ocrelizumab) for multiple sclerosis function through diverse mechanisms of action and are generally used in individuals who did not respond to other treatments. Mechanisms include lymphocyte maturation, lymphocyte trafficking and B cell depletion. |
|
• |
Immuno-modulators (DMF, teriflunomide) comprise a large portion of the multiple sclerosis market, including DMF, which had the highest sales among multiple sclerosis therapeutics in the United States in 2019. The two main drug within this class, DMF and teriflunomide, are weakly immunometabolic by mechanism of action. DMF increases Nrf2 while teriflunomide reduces T cell proliferation by targeting a mitochondrial enzyme. |
|
• |
Glatiramer acetate is a synthetic analogue of a main antigen in multiple sclerosis that functions through an inhibition of myelin-reactive T cells. While Copaxone, a glatiramer acetate, was the market-leading multiple sclerosis therapy in 2016, as of 2019 it sits outside of the top five brands for relapsing-remitting multiple sclerosis, with an approximate 7% market share. |
|
• |
Anti-trafficking agents (fingolimod) are among the newest therapies in multiple sclerosis and generally target S1P receptors to block the migration of lymphocytes. |
|
• |
Corticosteroids (methylprednisolone, dexamethasone) are commonly administered in multiple sclerosis intravenously in high doses over the period of three to five days in response to a disease flare. Efficacy in shortening disease flare is generally recognized. |
53
Limitations of these disease-modifying therapies include:
|
• |
Safety concerns. The profile of adverse effects of current therapeutics in multiple sclerosis is similar to those experienced in other autoimmune indications. Biologics used for multiple sclerosis increased the risk of infections and progressive multifocal leukoencephalopathy. Steroids and immuno-modulators result in liver toxicities and suppressed white and red blood cell counts. Anti-trafficking agents have been associated with cardiovascular complications and macular edema. With therapeutics that require chronic use, significant room for improvement is present in regards to safety. |
|
• |
Limited efficacy and loss of response. Across clinical trials of current therapeutics in relapsing-remitting multiple sclerosis, the rate of no evidence of disease activity has been observed to be 20% to 37% and patients with average age over 40 have experienced on average 25% or less inhibition of disability progression. The most recent approval for progressive multiple sclerosis was ocrelizumab, which offers less than a 7% effect size relative to placebo in 12-week disability progression. |
|
• |
Disability progression. Current therapeutics range from 10% to 45% of inhibition of disability progression. As the majority of therapeutics are focused entirely on changing the immune response, limited effects on the underlying pathways of neurodegeneration are addressed. Progressive forms of MS have an altered immunological profile relative to relapse-remitting multiple sclerosis, with innate immune responses predominating. Most multiple sclerosis therapeutics target the adaptive immune system to suppress flares. |
|
• |
Corticosteroid dependence. High-dose corticosteroids have short-term benefit in the resolution of disease flares. Chronic dependence on high-dose corticosteroids, even in pulses has been associated with cognitive deficiencies, depression, and remyelination and repair. Use in multiple sclerosis may be detrimental to long-term health. In neurological injuries and animal models of EAE, high-dose corticosteroids were associated with exacerbation of neuronal damage. |
Our solution for the treatment of multiple sclerosis
We believe the NX-66, through a differentiated NLRX1 pathway, if approved, may offer a competitive advantage relative to current therapies given its potential to address neurodegeneration and disease progression. NLRX1 is one of three regulatory NOD-like receptors (NLRs), in an overall family of 23 NLRs. Genetic abnormalities in NLR genes are linked to MS pathology, such as the Q705K polymorphism resulting in an overactive NLRP3 inflammasome and multiple mutations in NLRP1. NLRX1 is uniquely positioned as the natural counterbalance to these inflammatory NLRs that are overactive in multiple sclerosis. Preclinically, there is abundant evidence for the potency of NLRX1 in EAE through loss of function studies. In active (MOG immunization), passive (adoptive transfer) and spontaneous (2D2) models of EAE, the loss of NLRX1 leads to greater disease activity scores, increased number and severity of spinal cord lesions, and greater microglia activation. Additionally, NLRX1 contributes to the prevention of neurodegeneration through promoting controlled apoptosis over necrosis and pyroptosis, and regulating oxidative stress. Thus, we believe NLRX1 may aid in progressive forms of multiple sclerosis and address the therapeutic in multiple sclerosis based on the following observed characteristics:
|
• |
Tolerability. Preclinically, NLRX1 activation has not resulted in any dose-limiting toxicities in the study of NX-13 or NX-66. Mechanistically, NX-66 profiles most similarly to DMF out of current therapeutics. Importantly, DMF is often the choice of therapy when lack of side effects is a main point of concern by clinicians and patients. |
|
• |
Efficacy and loss of response. NLRX1 bridges innate and adaptive immune pathways, unlike other therapeutics. Progressive forms of MS have a notably different immune profile in the CNS, with an increased importance for microglia over CD4+ T cells in comparison to relapsing-remitting forms. NLRX1 is able to modulate both arms with effects directly on CD4+ T cells, through a down-regulation of Th1 and Th17 phenotypes, and microglia, through a promotion of repair pathways. We believe the ability to modulate both aspects of the disease course in multiple sclerosis may alleviate damage accrual. |
|
• |
Disability progression. We believe NLRX1 has the potential to directly prevent neuronal damage through control of oxidative stress and mitochondrial dysfunction in neurons, as well as promote apoptosis over necrosis. Disability progression results from an inability to remyelinate neurons and an imbalance in neuronal maintenance toward cell death. Limited therapies exist to address neuronal stress outside of immunomodulation. |
54
|
• |
Corticosteroid dependence. Providing an orally-active therapeutic for multiple sclerosis with limited side effects may enable greater patient adherence, earlier treatment with potent therapy, and an alternative mechanism for non-responders to current therapies. Combined, we believe these three items could decrease the use of corticosteroids. |
Preclinical results
Therapeutic dosing of oral NX-66 (20 mg/kg), between days 14 and 23 post-challenge, ameliorated disease severity in a MOG-induced model of EAE. NX-66 provided a greater than 50% reduction in disease activity four days after the initiation of treatment (n = 10, P ≤ 0.05). NX-66 treatment decreased Tnf and IL1b expression in the spinal cords of EAE mice.

Disease activity index in a MOG35-55 induced EAE model. Treatment with NX-66 (20 mg/kg, oral) was initiated at day 14 post-challenge and continued until day 23. TNF and IL-1b expression in the spinal cords of mice on day 23 (n = 10, P ≤ 0.05).
NX-66 exerts neuroprotective effects in N2A cells. In an MTT assay of viability, NX-66 treatment prevents loss of viability and cell damage in response to rotenone, an inducer of reactive oxygen species, and TNF.

Prevention of cell death in N2A cells stimulated with rotenone (100 µM) or TNF (100 ng/mL) by NX-66 (50, 100, 200 nM). Asterisks mark significance relative to vehicle (n = 9, P ≤ 0.05).
Preclinical GLP toxicology
NX-66 will be tested in a seven-day dose range-finding study in rats up to doses of 1,000 mg/kg in the second half of 2021. PK studies testing the bioavailability and profile of NX-66 in rats in the therapeutic window (1-20 mg/kg) will be conducted in the first half of 2021. GLP 28-day repeat dose toxicity studies in rats and dogs will be initiated in the second half of 2021.
Clinical development plan
Following an expected submission of an IND in 2022, we intend to initiate a Phase 1 clinical trial in normal healthy volunteers thereafter in the second half of 2022. This study will feature a standard single ascending dose/multiple ascending dose design testing a dose range 5-fold to 10-fold greater than projected therapeutic dose. The primary objective of the study will be to assess the safety and tolerability of NX-66 by frequency and severity of AEs, standard safety laboratory results, ECG and vital signs. The secondary objective will be to assess the PK of NX-66 in humans within blood and the CSF.
55
NX-66 for the treatment of Alzheimer’s disease
Overview
We are also developing NX-66, a small molecule product candidate targeting the NLRX1 pathway, for the treatment of Alzheimer’s disease. NLRX1 activation can protect neurons from oxidative stress and ameliorate CNS inflammation. We believe NX-66 may represent a novel approach to addressing an unmet clinical need in Alzheimer’s disease, for which no current therapy slows the progression of cognitive decline and neurological damage. The primary driver of NX-66’s development in Alzheimer’s disease is this unmet need, promising preclinical data and mechanistic alignment with the immunometabolic dysfunction present in disease. We intend to submit an IND for NX-66 in 2022.
Background on Alzheimer’s disease and current treatments
Alzheimer’s disease is an inflammatory and neurodegenerative disease that causes dementia. Alzheimer’s disease is a progressive disorder beginning with mild cognitive impairment and slight memory loss progressing into difficulty with everyday tasks and further into requirement of 24-hour care. Alzheimer’s disease affects up to 7 million patients in the United States with an estimated half million new cases diagnosed each year. With an expanding elderly population, the global therapeutics market for Alzheimer’s disease is estimated to be $12.9 billion by 2028 and is projected to have a compound annual growth rate of 19.3% between 2018 and 2028. With decreased quality of life and high home and hospice care costs, the total health care cost related to Alzheimer’s disease is estimated to be over $300 billion.
No current treatment for Alzheimer’s disease has been proven to slow the damage of neurons that is associated with cognitive decline. Four drugs are currently approved for the treatment of Alzheimer’s disease:
|
• |
Galantamine is an acetylcholinesterase inhibitor that targets amelioration of cholinergic deficiency. Galantamine can be isolated as a natural product and is generally used in the treatment of mild to moderate cases with reported effects on slight improvements in cognitive function and memory. |
|
• |
Donepezil is a cholinesterase inhibitor with primary mechanism of action tied to reversing the cholinergic deficiency present in Alzheimer’s disease. Donepezil provides a slight improvement in cognitive function without an effect on progression of disease. |
|
• |
Rivastigmine is an additional cholinesterase inhibitor that is used in the treatment of mild to moderate Alzheimer’s disease. As with other members of this drug class, its efficacy in the treatment of Alzheimer’s disease is primarily tied to symptomatic relief. |
|
• |
Memantine is a NMDA receptor antagonist that blocks excessive stimulation of neurons. Clinical trials of memantine resulted in no behavioral or neurological improvement, but a significant functional improvement, particularly in Severe Impairment Battery. Memantine and donepezil are also approved for use in combination. |
All four approved drugs provide symptomatic relief by targeting neurological signaling. Recent failures in clinical development in Alzheimer’s disease have largely focused on the plaques and tangles associated with disease. Mechanistically, limited late-stage clinical development of anti-inflammatory therapies has been conducted. Microglial activation has been linked to amyloid and tau pathologies and can contribute to neuronal cell death. Inflammation is also highly linked to metabolism with dysregulation of lipids and glucose resulting from chronic activation. Defects in cholesterol biosynthetic pathways are main genetic risk factors for Alzheimer’s disease. The brain during Alzheimer’s disease experiences many of the same deficiencies in glycemic control present in type 1 diabetes. The combined inflammatory, metabolic and oxidative stress results in a poor environment for restoring cognitive function.
Our solution for the treatment of Alzheimer’s disease
We believe NX-66, through a differentiated NLRX1 pathway, if approved, may offer a competitive advantage relative to current therapies given its immunometabolic pathway and potential to address neurodegeneration and disease progression. The immunopathology of Alzheimer’s disease shares common elements with progressive forms of multiple sclerosis, including overactivation of the inflammasome and the central nature of immunometabolism in the interaction between neurons and immune cells. The inflammasome contributes to the overproduction of IL-1ß and other inflammatory cytokines in response to the amyloids and fatty acids that accumulate in the central nervous system with age. Meanwhile, loss of NLRP3 protects against dementia in the APP/PS1 model and other models of Alzheimer’s disease. Aside from proteins directly linked to amyloid deposition, APP and PSEN1, many of the recently identified genetic variants associated with Alzheimer’s disease are rare receptors, such as ABCA7 or TREM2, which contribute to ATP levels and lipid homeostasis. NLRX1 activation can counteract both inflammasome activation and diminished cellular ATP.
56
In models of brain injury, loss of NLRX1 worsens disease pathology, increasing oxidative stress and promoting metabolic dysfunction. Activation of NLRX1 in vitro protects against cell death in the context of inflammatory and oxidative injury.
Preclinical GLP toxicology
We intend to initiate GLP IND-enabling studies for NX-66 in 2021.
Clinical development plan
We intend to initiate a safety and tolerability study for NX-66 in normal healthy volunteers in the second half of 2022.
NX-73 for the treatment of asthma
Overview
NX-73 is a small molecule product candidate that targets NLRX1 that we are developing for the treatment of Asthma. NLRX1 is a mitochondria-associated receptor involved in down-regulating inflammation during bacterial and viral exposure, colitis, multiple sclerosis and chronic pulmonary disease. Asthma encompasses a wide range of allergic and inflammatory diseases. Severe sub-types of asthma, including both neutrophilic and eosinophilic manifestations, lack effective treatment methods. We believe that NX-73, if approved, has the potential to improve on the current treatment options through the potential to resolve neutrophil inflammation, improve pulmonary function and reverse underlying fibrosis. We intend to submit an IND for NX-73 in the second half of 2023.
Background on asthma and current treatments
Over 25 million patients in the United States have asthma, representing a $14 billion annual therapeutic market that is expected to grow at a compound annual growth rate of 4% from 2019 to 2022. Multiple subtypes of asthma exist. Immunologically, these subtypes are dominated by a classification into type 2 and non-type 2. While most individuals with type 2 asthma will experience moderate response to standard treatments including corticosteroids, non-type 2 asthma, dominated by neutrophilic disease, is often refractory to these medications and results in severe, untreated disease in 10% to 15% of patients. The disease results in self-perpetuating disease in which airway epithelial cells are damaged and recruit neutrophils that chronically infiltrate the lung and contribute to epithelial cell damage. Limitations of current therapies for the treatment of asthma include:
|
• |
Failure to resolve neutrophilic inflammation. Corticosteroids, the main treatment for asthma, have been shown to promote the survival of neutrophils. Recent biologics and other treatments in development for severe eosinophilic asthma are focused on modulating Th2 and eosinophilic responses that leave Th17 and neutrophil inflammation unaffected. |
|
• |
Incompletely reversible airway obstruction. Neutrophilic asthma is associated with chronic airway obstruction requiring frequent intervention and severe health complications. This suggests that structural or fibrotic changes may be present that are not addressed with current therapies. |
|
• |
Refractory patients. Neutrophilic disease results in a greater likelihood that a patient will be refractory to current treatment. Refractory asthma patients tend to present with higher neutrophil counts. The noneosinophilic, or neutrophilic, asthma population is roughly half of the overall asthma population. |
Our solution for the treatment of asthma
Neutrophilic asthma is commonly associated with occupational and environmental molecules and is likely caused by unresolving neutrophil infiltration resulting from deregulated repair of the epithelium. NLRX1 is a receptor expressed both within pulmonary immune cells and airway epithelial cells. NLRX1 is responsible for maintaining mitochondrial metabolism in airway epithelial cells and preventing oxidative stress-induced cell death. In the absence of NLRX1, airway epithelial cells produce higher levels of neutrophil chemoattractants, such as IL-8, and Th17 polarizing cytokines, such as IL-6, in response to DAMPs and PAMPs. Immunologically, NLRX1 has been associated with altered responses to viral and fungal diseases of the lung. In both cases, NLRX1 was observed to be beneficial, lessening the severity of disease through the innate immune system. We believe NX-73, if approved, may address an unmet clinical need in refractory neutrophilic asthma by:
|
• |
Resolving neutrophil inflammation. NLRX1 activation reduces IL-8 and other neutrophil chemoattractants in airway epithelial cells. NLRX1 is a potent controller of CD4+ T cell differentiation through the downregulation of Th17 cells by inhibition of LDH. Both factors can decrease the recruitment of neutrophils to the lungs. |
57
|
• |
Improving epithelial cell health. Neutrophils can contribute to the death of epithelial cells through oxidative stress and metabolic dysfunction. NLRX1 activation increases the expression of antioxidant enzymes and also maintains mitochondrial metabolism. The restoration of the epithelial barrier integrity will improve pulmonary function. |
|
• |
Reversing underlying fibrosis. Pulmonary fibrosis may contribute to the inability to completely reverse airway obstruction. By resolving the neutrophil inflammation and decreasing TNF production, the activation and proliferation of fibroblasts will decrease, which can contribute to the improvement of fibrosis. |
Preclinical GLP toxicology
We intend to initiate a seven-day dose range finding study of NX-73 in rats up to doses of 1000 mg/kg, and PK studies testing the bioavailability and profile of NX-73 in rats in the therapeutic window (1-20 mg/kg) in the second half of 2021. We intend to initiate GLP 28-day repeat dose toxicity studies in rats and dogs in the second quarter of 2023.
Clinical development plan
Following an expected IND submission in the second half of 2023, we intend to initiate a Phase 1 study in normal healthy volunteers will be in the first half of 2024. The normal healthy volunteer study will feature a standard single ascending dose/multiple ascending dose design testing a dose range 5-fold to 10-fold greater than projected therapeutic dose. The primary objective of the study will be to assess the safety and tolerability of NX-73 by frequency and severity of AEs, standard safety laboratory results, ECG and vital signs. The secondary objective will be to assess the PK of NX-73 in humans.
NX-73 for the treatment of COPD
Overview
NX-73 is a small molecule product candidate that targets NLRX1 that we are developing for the treatment of COPD. NLRX1 is a mitochondria-associated receptor involved in down-regulating inflammation during bacterial and viral exposure, colitis, multiple sclerosis and chronic pulmonary disease. COPD is an inflammatory disease of the lung characterized by chronic bronchitis and emphysema and caused primarily by environmental exposures and cigarette smoke. The current treatments for COPD primarily provide symptomatic relief and slightly reduce severe flares and do not reduce the rate of progressive obstruction. We believe that NX-73 has the potential to improve on the current treatment options through the potential to address both epithelial and immune functions. We intend to submit an IND for NX-73 in the second half of 2023.
Background on COPD and current treatments
COPD is the third leading cause of death in the United States, may exist in up to 10% of adults over 40 and is estimated to be up to $12 billion annual market growing with a compound annual growth rate of nearly 6% from 2018 to 2028.
The primary class of treatment in COPD is bronchodilators including both short- and long-acting beta2 agonists and muscarinic antagonists. Inhaled corticosteroids are also used in certain cases, often in combination with methylxanthines, which improve the response to corticosteroids. The most recent new class approval in COPD was a PDE4 inhibitor, for which 80% of patients experienced moderate-to-severe COPD exacerbations, representing a 13% drop relative to placebo. PDE4 inhibition has struggled to penetrate the market, estimated to comprise 2% to 5%, attributable in part to mixed results during clinical development. Cytokine-based biologics have failed to improve respiratory function in clinical trials. Limitations of current therapies for the treatment of COPD include:
|
• |
Limiting effects on declining respiratory function. Current therapeutics for COPD primarily provide symptomatic relief and slightly reduce severe flares. Bronchodilators do not reduce the rate of progressive obstruction, in which patients have a higher likelihood of becoming refractory to treatment. This results in a limited impact on the overall mortality rate. |
|
• |
Failure to treat neutrophilic and severe disease. Bronchodilators provide little impact on the underlying inflammation present in COPD. While COPD patients with high eosinophil counts have partial response to corticosteroids, those without high eosinophils have limited response to any treatment. |
58
Our solution for the treatment of COPD
Alveolar macrophages are a highly plastic phenotype capable of maintaining tissue homeostasis and inducing robust inflammation in the lung. They are central players in COPD and chronic pulmonary diseases. NLRX1 is suppressed in human COPD and its expression correlates with the degree of airflow limitation. In cigarette smoke induced animal models, lack of NLRX1 results in emphysematous destruction and high expression of IL-18. Mechanistically, NLRX1 inhibits the PAMP and DAMP mediated activation of MAVS, a key effector protein for the induction of IL-18 and IL-1ß, as well as decrease oxidative stress tied to the damage of the airway epithelium and activation of alveolar macrophages.
COPD has a large refractory population and a lack of therapies that prevent further decline in severe disease. We believe a significant market opportunity exists for therapeutics addressing both epithelial and immune functions. NLRX1 activation can ameliorate alveolar destruction.
We believe NX-73, if approved, has the potential to address the lack of immune-based therapies. With the exception of PDE4 inhibitors, COPD treatment primarily addresses the symptoms without modulating the underlying inflammation. Similar to asthma, corticosteroids fail to improve disease in patients with high neutrophil counts. NLRX1 activation may reduce the infiltration of neutrophils through decreased Th17 cells and decreased IL-8 production by airway epithelial cells.
Preclinical GLP toxicology
We intend to initiate GLP IND-enabling studies for NX-73 in the second half of 2023.
Clinical development plan
We intend to initiate a safety and tolerability study in normal healthy volunteers of NX-73 in the first half of 2024.
Our PLXDC2 pathway product candidate
The PLXDC2 pathway
PLXDC2 is a transmembrane receptor associated with immunoregulatory functions. Immunologically, PLXDC2 activation leads to the production of IL-10 and prevention of oxidative stress. PLXDC2 intercepts the receptor for advanced glycation end-products (RAGE) signaling pathway. This leads to downstream inhibition of NF-κB and HIF-1α signaling that is associated with TNFα, IL-6, MCP1 and other cytokines. Further supporting this anti-inflammatory cascade is an inhibition of the semaphorin 4A/plexin B1 axis that regulates MAPK activation. Through proteolysis of the VEGF receptor, activation of PLXDC2 can also induce anti-angiogenic effects that can limit immune cell infiltration and defects in tissue vascularization and anti-fibrotic effects that can reduce the production of fibronectin.

59
|
•PLXDC2 activation leads to immunoregulatory functions •Production of IL-10 •Prevention of oxidative stress •Inhibition of NF-kB and HIF-1α signaling that decrease production of TNF, IL-6 and MCP-1 •Intercepts RAGE signaling pathway •Through proteolysis of the VEGF receptor activation of PLXDC2 can lead to: •Anti-angiogenic effects that limit immune cell infiltration •Anti-fibrotic effects that decrease fibronectin production |
|

The primary immune cell type affected by PLXDC2 activation is considered to be macrophages. When stimulated, PLXDC2 activated macrophages more closely associate with a non-inflammatory phenotype categorized by high expression of Retnla and Arg1 and secretion of IL-10. While macrophages are central to the effects of PLXDC2 activation, effects are also observed in adaptive immunity. In animal models, PLXDC2 activation is associated with decreased B cell maturation and lower local Th17 activation due in part to the inhibition of MAPK signaling and glycolysis that lessens proliferation. In addition to effects on the growth factor microenvironment, PLXDC2 activation is also associated with growth arrest in fibroblasts leading to decreased proliferation. This could offer the potential to halt tissue fibrosis that leads to deterioration of organ function.
The development of PLXDC2 focuses on diseases with implication of oxidative stress, fibrosis and angiogenesis. PLXDC2 is a highly relevant target for diabetic complications, such as nephropathy and retinopathy, due to the high levels of advanced glycation end-products that result from hyperglycemia. These indications, in addition to rheumatoid arthritis, are implicated with defects in local growth factor production and innate immune cytokines that may be improved with PLXDC2 activation.
PX-69 for the treatment of diabetic nephropathy
Overview
PX-69 is a small molecule product candidate that targets PLXDC2 that we are developing for the treatment of diabetic nephropathy. Diabetic nephropathy is a main complication in both type 1 and type 2 diabetes and is the leading cause of ESRD. While current treatments are effective as a preventative measure, they are less efficacious when initiated at later stages of disease. We believe that PX-69 has the potential to improve on the current treatment options through the potential to improve kidney function and address late stage renal disease. We intend to submit an IND for PX-69 in the first half of 2022.
Background on diabetic nephropathy and current treatments
Forty percent of diabetic patients will develop diabetic nephropathy, affecting both type 1 and type 2 diabetics and resulting in up to 10 million patients in the United States. Diabetic nephropathy is the leading cause for long-term renal dialysis and ESRD. The estimated global market for diabetic nephropathy is up to $3 billion annually with a compound annual growth rate of over 5% from 2014 to 2020.
Prolonged periods of hyperglycemia and dysregulated glucose metabolism increases the risk for many complications, from kidney failure to blindness to amputation of extremities. High blood glucose results in the build-up of multiple metabolites that can directly damage the kidney and cause oxidative stress. Patients often require high blood pressure- and cholesterol-lowering drugs and dialysis resulting from these complications.
60
Currently, pharmacological management of diabetic nephropathy is managed through glycemic control and blood pressure medications. The most commonly prescribed treatments for diabetic nephropathy are angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blocker (ARB). These treatments are believed to function through improvement of a hemodynamic imbalance, reducing stress on the kidneys and overall inflammation. While effective as a preventative measure when early detection is possible, diminished efficacy of these therapies is observed when initiated at later stages of disease. These therapies unreliably decrease serum creatinine indicating that further improvement on removal of waste products is possible.
Our solution for the treatment of diabetic nephropathy
The native protein ligand of PLXDC2 is locally expressed in both the retina and kidney and is notably suppressed when damage to either organ occurs. In the kidney, the presence of this protein is associated with suppression of advanced glycation end-product signaling and inhibition of fibrosis. Many diabetic complications result from advanced glycation end-products which are formed from the reaction of high glucose levels with proteins and other macromolecules. By intercepting RAGE signaling, PLXDC2 prevents myriad downstream effects that lead to kidney damage. Primary among these mechanisms is the prevention of oxidative stress and high-glucose induced activation of NF-κB and HIF1α. Activation of PLXDC2 results in the downregulation of TNF, MCP1 and other cytokines. Inhibition of fibrosis, including a downregulation of fibronectin, is caused by inhibition of RAGE combined with VEGF receptor proteolysis. The second of these effects also serves to reduce angiogenesis, an additional process connected to diabetic nephropathy. Enhanced activation of PLXDC2 may therefore help return the tissue to homeostasis in the deficiency of the native ligand.
We believe PX-69, if approved, has the potential to improve on current therapies by:
|
• |
Providing direct antioxidant effects. PLXDC2 activation is associated with decreased NF-κB activation and lower intracellular reactive oxygen species. Combined with reduction of local hypoxia, PLXDC2 can improve the health and homeostasis of renal epithelial cells and podocytes. |
|
• |
Addressing late-stage disease. PLXDC2 has the potential to intercept the angiogenic and fibrogenic signals that are more prevalent during late-stage disease. In the absence of improved screening, patients with stage 4 or higher renal disease will continue to have a higher likelihood of requiring long-term dialysis or kidney transplant. |
|
• |
Improvement of overall kidney function. The build-up of multiple waste products in the body can result in additional co-morbidities that contribute to the overall decline in health in diabetic individuals. Identification of novel treatment strategies in diabetic nephropathy may help to reduce these in a wider range of individuals. |
Preclinical GLP toxicology
PX-69 will be tested in a seven-day dose range finding study in rats up to doses of 1,000 mg/kg in the second quarter of 2021. We intend to initiate PK studies testing the bioavailability and profile of PX-69 in rats in the therapeutic window (1-20 mg/kg) in the second quarter of 2021. We intend to initiate GLP 28-day repeat dose toxicity studies in rats and dogs in the fourth quarter of 2022.
Clinical development plan
Following an expected IND submission in the first half of 2022, we intend to initiate a Phase 1 study in normal healthy volunteers in the second half of 2022. The normal healthy volunteer study will feature a standard single ascending dose/multiple ascending dose design testing a dose range 5-fold to 10-fold greater than projected therapeutic dose. The primary objective of the study will be to assess the safety and tolerability of PX-69 by frequency and severity of AEs, standard safety laboratory results, ECG and vital signs. A secondary objective will be to assess the PK of PX-69 in humans.
PX-69 for the treatment of rheumatoid arthritis
Overview
PX-69 is a small molecule product candidate that targets PLXDC2 that we are developing for the treatment of rheumatoid arthritis. Rheumatoid arthritis is characterized by a swelling and loss of mobility in joints caused by excessive inflammation and infiltration into the joint synovium. There is no clear advantage in terms of safety and efficacy for the current approved treatments. We believe that PX-69 has potential to improve on the modest efficacy observed with current treatments in rheumatoid arthritis due to disease-specific and general immune functions. We intend to submit an IND for PX-69 in the first half of 2022.
61
Background on rheumatoid arthritis and current treatments
Rheumatoid arthritis is a global health concern and affects approximately 1.3 million patients in the United States and represents an approximate $25 billion annual therapeutics market. The market is estimated to be the second largest specialty drug indication behind oncology.
Five classes of drugs (NSAIDs, corticosteroids, DMARDs, biologics and JAK inhibitors) are used to manage rheumatoid arthritis. The rheumatoid arthritis market is fragmented due to no clear advantage in terms of safety and efficacy among these classes of therapies. The result is a lack of a defined treatment paradigm or order of escalation between classes.
Our solution for the treatment of rheumatoid arthritis
Rheumatoid arthritis causes severe inflammation of joints leading to loss of mobility and intense pain. The underlying immunology of the synovial inflammation is complex involving the interplay of myeloid cells, T cells, fibroblasts and other structural cells of the synovium. High expression of TNF and IL-6 are central to the pathogenesis of rheumatoid arthritis, with additional contributions by IL-1ß, IL-12, IL-17, IL-21, IL-23, MCP1, and TGF-ß. Together these cytokines can lead to leukocytic recruitment, bone remodeling, pannus formation, oxidative stress and hyperplasia of the joint lining. As a strong regulator of myeloid responses, including the production of TNF and IL-6 as well as overall infiltration and angiogenesis, PLXDC2 can serve as a novel target in rheumatoid arthritis.
Rheumatoid arthritis results from interactions between immune cells and synoviocytes that leads to joint swelling, pannus formation and bone loss. These interactions are exacerbated by increased angiogenesis driven by local hypoxia. PLXDC2 is a receptor for an anti-angiogenesis factor that limits angiogenesis through proteolysis of the VEGF receptor. The semaphorin 4A/plexin B1 axis is a major contributor to interactions between Th17 cells and synovial fibroblasts. The axis leads to downstream upregulation of MAPK and production of factors contributing to osteoclastogenesis. PLXDC2 is an inhibition of this pathway and MAPK activation.
We believe PX-69, if approved, has the potential to improve on the modest efficacy observed with current treatments in rheumatoid arthritis due to disease-specific and general immune functions. PLXDC2 has a greater potential to influence osteoclastogenesis and angiogenesis than current therapeutics from a mechanistic standpoint. If combined with high tolerability, we believe PX-69 could be a disruptive therapy in rheumatoid arthritis.
Preclinical GLP toxicology
We intend to initiate GLP IND-enabling studies for PX-69 in the second quarter of 2021.
Clinical development plan
We intend to initiate a safety and tolerability study for PX-69 in normal healthy volunteers in the second quarter of 2022.
Manufacturing
Our drug substance and drug product manufacturing are conducted at third-party contract manufacturing organizations, or CMOs, in India and Switzerland. All of our manufacturers hold applicable licenses, certifications and/or approvals for cGMP manufacturing, analytical testing, packaging, and release operations from multiple drug regulation entities, including the FDA. Each manufacturer has also been independently qualified through our own internal qualification processes.
Multiple batches of BT-11 drug substance and drug product candidate have been completed with completed two-year room temperature stability data. BT-11 drug product is an immediate release tablet at multiple strengths. Process optimization, scale-up, manufacturing and production of registration batches are expected in 2021. Manufacture of NX-13 drug substance and drug product has been completed for Phase 1 studies with indications of room temperature stability in all stability assessments as of August 2020. NX-13 drug product candidate is an immediate release tablet at multiple strengths. Process optimization and scale-up for Phase 2 clinical trial supply will occur in 2021. Phase 3 process optimization, scale-up and registration batch manufacture are projected for completion in 2023.
62
We have recently begun the process of expanding our manufacturing capacity by developing relationships with new, qualified CMOs in other parts of the world. Additional CMOs for the manufacture of BT-11 and NX-13 drug substance and drug products are currently being identified, with each required to comply with current good manufacturing practices, or cGMP, and have the relevant manufacturing expertise for the applicable product. We expect that the newly qualified CMOs will begin process transfer and scale-up activities by the end of 2020 with plans to begin cGMP clinical manufacture in 2021. Together with our current manufacturing capabilities, this added capacity and strategic placement is expected to satisfy the regulatory demands of each of the anticipated long-term commercial markets while also ensuring a continuance of supply.
Competition
The biotechnology and pharmaceutical industries, and particularly the market for the treatment of autoimmune diseases, are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary and novel products and product candidates. We face competition with respect to our current product candidates, and will face competition with respect to any product candidates that we may seek to develop or commercialize in the future, from many different sources, including major pharmaceutical and specialty pharmaceutical companies, compounding facilities, academic institutions and governmental agencies and public and private research institutions.
We are aware of several other products and product candidates as potential treatments for UC and Crohn’s disease that would compete with BT-11 and NX-13, if approved. In particular, we expect to compete against companies that produce biologic drugs that currently dominate the UC and Crohn’s disease market, as well as companies that produce the aminosalicylates, steroids and immunosuppressants that are currently used to treat patients with mild to moderate disease. If approved, BT-11 and NX-13 are expected to compete against companies that produce, or are developing, injectable biologic therapeutics such as AbbVie Inc., Eli Lilly and Co., Janssen Pharmaceuticals, Inc., Roche Holding Ltd., Takeda Pharmaceutical Company Ltd. and UCB S.A., as well as companies that produce, or are developing, oral products such as Applied Molecular Transport Inc., Arena Pharmaceuticals, Inc., Bristol-Myers Squibb Co., Galapagos N.V., Gilead Sciences, Inc., Gossamer Bio, Inc., Pfizer Inc., Protagonist Therapeutics, Inc. and Theravance Biopharma, Inc.
BT-11 and NX-13 are well differentiated orally active, gut-restricted, therapeutic candidates that are the first to target their respective pathways. We are not aware of any product candidate targeting the LANCL2, NLRX1, or PLXDC2 pathways in current clinical development.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer or more effective, have fewer or less severe side effects, are more convenient or are less expensive than BT-11, NX-13 or any other product that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for our product, which could result in our competitors establishing a strong market position before we are able to enter the market.
Many of the companies against which we are competing, or against which we may compete in the future, have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or that may be necessary for, our programs. We believe that our unique corporate culture, proprietary LANCE platform, and well-differentiated technology will help us effectively navigate the challenges associated with this competitive landscape targeting a large market of over $153 billion by 2025.
63
Intellectual property
Patents and applications
Our success depends in part on our ability to obtain and maintain proprietary protection for our product candidates, technology, and know-how; to operate without infringing the proprietary rights of others; and to prevent others from infringing our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, pursuing and obtaining patent protection in the United States and jurisdictions outside the United States. Our patent portfolio is intended to cover our product candidates, their methods of use, and any other inventions that are commercially important to our business. We also rely on trade secret protection of our confidential information and know-how relating to our proprietary technology, platforms, and product candidates.
BT-11 patent portfolio
We solely own a patent portfolio covering the BT-11 compound and its use in treating inflammatory bowel disease (e.g., UC and CD) and other conditions.
The BT-11 patent portfolio includes five issued U.S. patents: U.S. Patent Nos. 9,556,146; 9,839,635; 10,028,950; 10,682,349; and 10,849,895. These patents collectively cover the BT-11 compound from a variety of angles and degrees of breadth. These patents also collectively cover compositions containing BT-11 and methods of using BT-11 for treating inflammatory bowel disease and other conditions.
The BT-11 portfolio also includes issued patents covering BT-11 in Australia (AU Patent No. 2015337091), China (CN Patent No. 107108573), Europe (EP Patent No. 3209655), Hong Kong (HK Patent No. 1240921), Israel (IL Patent No. 252630), Japan (JP Patent Nos. 6499306 and 6806829), New Zealand (NZ Patent No. 732213), the Republic of Korea (KR Patent No. 10-2026342), and the Russian Federation (RU Patent No. 2688677). The European patent is validated in each of Albania, Austria, Belgium, Bulgaria, Croatia, the Czech Republic, Denmark, Finland, France, Germany, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxemburg, Macedonia, Malta, Monaco, the Netherlands, Norway, Poland, Portugal, Romania, Serbia, Slovenia, Spain, Sweden, Switzerland, Turkey, and the United Kingdom. The issued Australian and Russian patents further cover methods of using BT-11 for treating inflammatory bowel disease and other conditions.
The BT-11 portfolio also includes pending applications in Brazil, Canada, India, and Turkey.
The application lineages in all the aforementioned jurisdictions remain pending, which provides the opportunity to pursue protection of additional aspects of BT-11 and/or its uses.
The issued patents and any patents issuing from the pending applications in this portfolio are projected to expire in 2035, absent any surrendered term, adjustments, or extensions.
NX-13 patent portfolio
We solely own a patent portfolio covering the NX-13 compound and its use in treating inflammatory bowel disease and other conditions.
The NX-13 patent portfolio includes two issued U.S. patents: U.S. Patent Nos. 10,487,057 and 10,676,436. These patents collectively cover the NX-13 compound and the use of NX-13 in treating inflammatory bowel disease and other conditions. A continuation application to pursue additional coverage of NX-13 and its uses is pending.
The NX-13 patent portfolio also includes pending applications in Argentina, Australia, Brazil, Canada, Chile, China, Eurasia, Europe, Georgia, India, Israel, Japan, Mexico, New Zealand, the Republic of Korea, Ukraine, and Uzbekistan.
The issued patents and any patents issuing from the pending applications in this portfolio are projected to expire in 2039, absent any surrendered term, adjustments, or extensions.
BT-11-conditioned T cells
We solely own a patent application portfolio pursuing coverage of BT-11-conditioned T cells, methods of making the T cells, and methods of using the T cells in treating inflammatory bowel disease and other conditions. This portfolio includes pending applications in the U.S., Australia, Brazil, Canada, Chile, China, Eurasia, Europe, Georgia, Hong Kong, India, Israel, Japan, Mexico, New Zealand, the Republic of Korea, Ukraine, and Uzbekistan. Any patents issuing from these applications are predicted to expire in 2038, absent any surrendered term, adjustments, or extensions.
64
Other LANCL2 targeting compound patent portfolio
We solely own a patent portfolio covering other LANCL2-targeting compounds and their use in treating inflammatory bowel disease and other conditions. This portfolio includes issued patents in the United States (U.S. Patent Nos. 10,201,538 and 10,493,072), the Republic of Korea (KR Patent No. 10-2134171), and the Russian Federation (RU Patent No, 2741576) and pending applications in the United States, China, Israel, Japan, and New Zealand. The issued patents and any patents issuing from the pending applications in this portfolio are projected to expire in 2035, absent any surrendered term, adjustments, or extensions.
BT-104
We solely own a patent application portfolio pursuing coverage of the BT-104 compound, compounds related to BT-104, and uses of such compounds. This portfolio includes a pending U.S. application and a pending PCT application that is eligible for worldwide filing. Any patents issuing in this portfolio are projected to expire in 2040, absent any surrendered term, adjustments, or extensions.
PX-69
We solely own a patent application portfolio pursuing coverage of the PX-69 compound, compounds related to PX-69, and uses of such compounds. This portfolio includes pending U.S., Argentina, and PCT applications. Any patents issuing in this portfolio are projected to expire in 2041, absent any surrendered term, adjustments, or extensions.
NX-66
We solely own a U.S. provisional patent application directed to the NX-66 compound, compounds related to NX-66, and uses of such compounds. We plan to file U.S. and PCT nonprovisional applications claiming priority to this provisional application. If the nonprovisional applications are timely filed and granted, the granted patents would expire in 2041, absent any surrendered term, adjustments, or extensions.
BT-111
We solely own a U.S. provisional patent application directed to the BT-111 compound, compounds related to BT-111, and uses of such compounds. We plan to file U.S. and PCT nonprovisional applications claiming priority to this provisional application. If the nonprovisional applications are timely filed and granted, the granted patents would expire in 2041, absent any surrendered term, adjustments, or extensions.
BT-11 polymorphs
We solely own a U.S. provisional patent application directed to crystalline forms of BT-11 and uses of such crystalline forms. We plan to file U.S. and PCT nonprovisional applications claiming priority to this provisional application. If the nonprovisional applications are timely filed and granted, the granted patents would expire in 2041, absent any surrendered term, adjustments, or extensions.
Topical BT-11 therapies
We solely own a U.S. provisional patent application directed to the topical administration of BT-11 and related compounds for the therapeutic treatment of psoriasis and other conditions of the skin and mucosa. We plan to file U.S. and PCT nonprovisional applications claiming priority to these provisional applications. If the nonprovisional applications are timely filed and granted, the granted patents would expire in 2041, absent any surrendered term, adjustments, or extensions.
Additional patent applications
We plan to file U.S. provisional patent applications covering our NX-73 compound and its therapeutic uses within the next several months.
65
Intellectual Property Protection
We cannot predict whether the patent applications we pursue will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide any proprietary protection from competitors. Further, any issued patents may expire before the expected expiration dates disclosed above due to actions taken during patent prosecution, such as submission of a disclaimer surrendering the term of a patent beyond a certain date. Even if our pending patent applications are granted as issued patents, those patents, as well as any patents we license from third parties, may be challenged, circumvented, or invalidated by third parties. While there are currently no contested proceedings or third-party claims relating to any of the patents or patent applications described above, we cannot provide any assurances that we will not have such proceedings or third-party claims at a later date or once any patent is granted.
The term of a patent depends upon the legal term of patents in the particular country in which it is obtained. In most countries in which we file, including the United States, the patent term is 20 years from the earliest date of filing a non-provisional patent application.
In the United States, the term of a patent that covers an FDA-approved drug may be eligible for patent term extension, which permits in some cases restoration of patent term as compensation for patent term lost during the FDA regulatory review process. In certain circumstances, the Hatch-Waxman Act permits a patent term extension of up to five years beyond the unextended expiration date of the U.S. patent. The length of the patent term extension is related to the length of time the approved drug is under regulatory review. Patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and only one patent applicable to an approved drug may be extended. Provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug, or provide an additional period of protection for the approved pharmaceutical product following expiry of the patent. In the future, if our products receive FDA approval, we expect to apply for patent term extensions on patents covering those products. We plan to seek patent term extensions to any of our issued patents in any jurisdiction where these are available. There is no guarantee, however, that the applicable authorities, including the U.S. Patent and Trademark Office in the United States and the national patent offices in Europe or other jurisdictions, will agree with our assessment of whether such extensions should be granted, and, if granted, the length of such extensions.
In addition to our reliance on patent protection for our inventions, product candidates, and research programs, we also rely on trade secret protection for our confidential and proprietary information. Although we take steps to protect our proprietary information and trade secrets, including through contractual means with our employees and consultants, third parties may independently develop substantially equivalent proprietary information and techniques, or otherwise gain access to our trade secrets or disclose our technology. Thus, we may not be able to meaningfully protect our trade secrets. It is our policy to require our employees, consultants, outside scientific collaborators, sponsored researchers, and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information concerning our business or financial affairs developed or made known to the individual or entity during the course of the party’s relationship with us is to be kept confidential, and not disclosed to third parties except in specific circumstances. In the case of employees, the agreements provide that all inventions conceived by the individual, and which are related to our current or planned business or research and development or made during normal working hours, on our premises or using our equipment or proprietary information, are our exclusive property. In addition, we take other appropriate precautions, such as physical and technological security measures, to guard against misappropriation of our proprietary technology by third parties. We have also adopted policies and conduct training that provides guidance on our expectations and practices to protect our trade secrets.
Freedom to Operate
We have obtained legal opinions on the freedom to operate with BT-11 and NX-13. The legal opinions have concluded that we have freedom to operate with BT-11, NX-13, and their uses in the treatment of inflammatory bowel diseases, such as UC and CD.
66
Government regulation
The FDA and comparable regulatory authorities in state and local jurisdictions and in other countries impose substantial and burdensome requirements upon companies involved in the clinical development, manufacture, marketing and distribution of drugs, such as those we are developing. These agencies and other federal, state and local entities regulate, among other things, the research and development, testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion, distribution, post-approval monitoring and reporting, sampling and export and import of our product candidates.
U.S. government regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and its implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA’s refusal to approve pending NDAs, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
|
• |
completion of pre-clinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s good laboratory practices, or GLP, regulations; |
|
• |
submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may begin; |
|
• |
approval by an independent institutional review board, or IRB, at each clinical site before each trial may be initiated; |
|
• |
performance of adequate and well-controlled human clinical trials in accordance with good clinical practice, or GCP, requirements to establish the safety and efficacy of the proposed drug product for each indication; |
|
• |
submission to the FDA of a New Drug Application, or NDA; |
|
• |
satisfactory completion of an FDA advisory committee review, if applicable; |
|
• |
satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current good manufacturing practices, or cGMP, requirements and to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
|
• |
FDA review and approval of the NDA. |
Pre-clinical studies
Pre-clinical studies include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies to assess potential safety and efficacy. An IND sponsor must submit the results of the pre-clinical tests, together with manufacturing information, analytical data and any available clinical data or literature, among other things, to the FDA as part of an IND. Some pre-clinical testing may continue even after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
67
Clinical trials
Clinical trials involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND (or equivalent submission ex-US). In addition, an IRB or ethics committee, or EC, at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their www.clinicaltrials.gov website.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined:
|
• |
Phase 1: The drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness. |
|
• |
Phase 2: The drug is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
|
• |
Phase 3: The drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB or EC can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Marketing approval
Assuming successful completion of the required clinical testing, the results of the pre-clinical and clinical studies, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. In most cases, the submission of an NDA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act, or PDUFA, guidelines that are currently in effect, the FDA has a goal of ten months from the date of “filing” of a standard NDA for a new molecular entity to review and act on the submission. This review typically takes twelve months from the date the NDA is submitted to FDA because the FDA has approximately two months to make a “filing” decision.
In addition, under the Pediatric Research Equity Act of 2003, or PREA, as amended and reauthorized, certain NDAs or supplements to an NDA must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements.
The FDA also may require submission of a risk evaluation and mitigation strategy, or REMS, plan to ensure that the benefits of the drug outweigh its risks. The REMS plan could include medication guides, physician communication plans, assessment plans, and/or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
68
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality and purity.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical trial sites to assure compliance with GCP requirements.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA and may require additional clinical or pre-clinical testing in order for FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Special FDA expedited review and approval programs
The FDA has various programs, including Fast Track designation, Breakthrough Therapy designation, Accelerated Approval, and Priority Review, which are intended to expedite or simplify the process for the development and FDA review of drugs that are intended for the treatment of serious or life-threatening diseases or conditions and demonstrate the potential to address unmet medical needs. The purpose of these programs is to provide important new drugs to patients earlier than under standard FDA review procedures.
Under the fast track program, the sponsor of a new drug candidate may request that FDA designate the drug candidate for a specific indication as a fast track drug concurrent with, or after, the filing of the IND for the drug candidate. Fast track designation provides opportunities for frequent interactions with the FDA review team to expedite development and review of the product. FDA may initiate review of sections of a fast track drug’s NDA before the application is complete. This rolling review is available if the applicant provides, and FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, FDA’s time period goal for reviewing an application does not begin until the last section of the NDA is submitted.
69
In addition, a sponsor can request breakthrough therapy designation for a drug if it is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs designated as breakthrough therapies are eligible for intensive guidance from FDA on an efficient drug development program, organizational commitment to the development and review of the product including involvement of senior managers, and, like fast track products, are also eligible for rolling review of the NDA. Both fast track and breakthrough therapy products are also eligible for accelerated approval and/or priority review, if relevant criteria are met.
Under the FDA’s accelerated approval regulations, the FDA may approve a drug for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions, or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A drug candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, will allow FDA to withdraw the drug from the market on an expedited basis. All promotional materials for drug candidates approved under accelerated approval regulations are subject to prior review by FDA.
Once an NDA is submitted for a product intended to treat a serious condition, the FDA may assign a priority review designation if FDA determines that the product, if approved, would provide a significant improvement in safety or effectiveness. A priority review means that the goal for the FDA to review an application is six months, rather than the standard review of ten months under current PDUFA guidelines. Under the current PDUFA agreement, these six- and ten- month review periods are measured from the 60-day filing date rather than the receipt date for NDAs for new molecular entities, which typically adds approximately two months to the timeline for review from the date of submission. Most products that are eligible for fast track breakthrough therapy designation are also likely to be considered appropriate to receive a priority review.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened. In addition, the manufacturer of an investigational drug for a serious or life-threatening disease is required to make available, such as by posting on its website, its policy on responding to requests for expanded access. Furthermore, fast track designation, breakthrough therapy designation, and priority review do not change the standards for approval and may not ultimately expedite the development or approval process.
Orphan designation
The FDA may grant orphan designation to drugs or biologics intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the United States, or if it affects more than 200,000 individuals in the United States, and there is no reasonable expectation that the cost of developing and marketing the product for this type of disease or condition will be recovered from sales in the United States. Orphan designation must be requested before submitting a NDA or Biologics License Application, or a BLA. After the FDA grants orphan designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
In the United States, orphan designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. In addition, if a product receives the first FDA approval for the indication for which it has orphan designation, the product is entitled to orphan exclusivity, which means the FDA may not approve any other application to market the same product for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority over the product with orphan exclusivity or where the manufacturer with orphan exclusivity is unable to assure sufficient quantities of the approved orphan designated product. Competitors, however, may receive approval of different products for
70
the indication for which the orphan product has exclusivity or obtain approval for the same product but for a different indication for which the orphan product has exclusivity. Orphan product exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval of the same product as defined by the FDA or if our product candidate is determined to be contained within the competitor’s product for the same indication or disease. If a drug or biological product designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan product exclusivity.
Post-approval requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There also are continuing, annual program user fee requirements for any marketed products, as well as application fees for supplemental applications with clinical data.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP requirements and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
|
• |
restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
|
• |
safety alerts, Dear Healthcare Provider letters, press releases or other communications containing warning or other safety information about the product; |
|
• |
fines, warning letters or holds on post-approval clinical trials; |
|
• |
refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product approvals; |
|
• |
product seizure or detention, or refusal to permit the import or export of products; or |
|
• |
injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act, or PDMA, which regulates the distribution of drugs and drug samples at the federal level, and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution.
71
Coverage and reimbursement
Sales of our product candidates, if approved, will depend, in part, on the extent to which the cost of such products will be covered and adequately reimbursed by third-party payors, such as government healthcare programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly limiting coverage and/or reducing reimbursements for medical products and services by challenging the prices and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. If these third-party payors do not consider our products to be cost-effective compared to other therapies, they may not cover our products after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
There is no uniform policy requirement for coverage and reimbursement for drug products among third-party payors in the United States. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. The coverage determination process can be a time-consuming and costly process that may require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be obtained or applied consistently. Even if reimbursement is provided, market acceptance of our products may be adversely affected if the amount of payment for our products proves to be unprofitable for healthcare providers or less profitable than alternative treatments, or if administrative burdens make our products less desirable to use.
In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our product candidates or a decision by a third-party payor to not cover our product candidates could reduce physician usage of our products candidates, once approved, and have a material adverse effect on our sales, results of operations and financial condition.
U.S. healthcare reform
There have been and continue to be proposals by the federal government, state governments, regulators and third-party payors to control or manage the increased costs of healthcare and, more generally, to reform the U.S. healthcare system. By way of example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, collectively referred to as the ACA, enacted in March 2010, has had and is expected to continue to have a significant impact on the healthcare industry. The ACA, among other things, imposed a significant annual fee on certain companies that manufacture or import branded prescription drug products, and established a new Medicare Part D coverage gap discount program, in which manufacturers must now agree to offer 70% point of-sale discounts off negotiated prices of applicable brand drugs and therapeutic biologics to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs and therapeutic biologics to be covered under Medicare Part D. The ACA also increased the Medicaid rebate rate and expanded the rebate program to include Medicaid managed care organizations. It also contained substantial new provisions intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for the healthcare industry, impose new taxes and fees on pharmaceutical manufacturers, and impose additional health policy reforms, any or all of which may affect our business.
There have been executive, judicial and Congressional challenges to certain aspects of the ACA. For example, the Tax Cuts and Jobs Act of 2017, or the Tax Act, included a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. Additionally, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and, effective January 1, 2021, also eliminated the health insurance tax. On December 14, 2018, a Texas U.S. District Court Judge ruled that the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress as part of the Tax Act. Additionally, on December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the District Court ruling that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. The United States Supreme Court is currently reviewing this case, but it unknown when a decision will be reached. Although the Supreme Court has not yet ruled on the constitutionality of the ACA, on January 28, 2021, President Biden issued an executive order to initiate a special enrollment period from February 15, 2021 through May 15, 2021 for purposes of obtaining health insurance coverage through the ACA marketplace. The executive order also instructs certain governmental agencies to review and reconsider their
72
existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA. It is unclear how the Supreme Court ruling, other such litigation, and the healthcare reform measures of the Biden administration will impact the ACA and our business.
Other legislative changes have also been proposed and adopted since the ACA was enacted. For example, the Budget Control Act of 2011 resulted in aggregate reductions in Medicare payments to providers of 2% per fiscal year, which went into effect in 2013 and, following passage of subsequent legislation, will stay in effect through 2030, with the exception of a temporary suspension from May 1, 2020 through March 31, 2021, unless additional Congressional action is taken. Additionally, the American Taxpayer Relief Act of 2012, among other things, reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. New laws may result in additional reductions in Medicare and other healthcare funding.
Further, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products. Such scrutiny has resulted in several recent Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products. At the federal level, the Trump administration used several means to propose or implement drug pricing reform, including through federal budget proposals, executive orders and policy initiatives. For example, on July 24, 2020 and September 13, 2020, President Trump announced several executive orders related to prescription drug pricing that attempt to implement several of the Administration’s proposals. The FDA also recently released a final rule, effective November 30, 2020, implementing a portion of President Trump’s Executive Order announced on July 24, 2020 that directed HHS to finalize the Canadian drug importation proposed rule previously issued by HHS and make other changes allowing for personal importation of drugs from Canada. The FDA final rule provides guidance for states to build and submit importation plans for drugs from Canada. Further, on November 20, 2020, HHS finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The implementation of the rule has been delayed by the Biden administration from January 1, 2022 until January 1, 2023 in response to ongoing litigation. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harbor for fixed fees to pharmacy benefit managers for certain services rendered to manufacturers, the implementation of which have also been delayed pending review by the Biden administration until January 1, 2023. On November 20, 2020, CMS issued an interim final rule implementing President Trump’s Most Favored Nation executive order, which would tie Medicare Part B payments for certain physician-administered drugs to the lowest price paid in other economically advanced countries, effective January 1, 2021. On December 28, 2020, the United States District Court in Northern California issued a nationwide preliminary injunction against implementation of the interim final rule. At the state level, legislatures have become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. It is also possible that additional governmental action is taken in response to the COVID-19 pandemic.
It is uncertain whether and how future legislation, whether domestic or foreign, could affect prospects for our product candidates or what actions foreign, federal, state, or private payors for healthcare treatment and services may take in response to any such healthcare reform proposals or legislation, particularly in light of the recent U.S. presidential election. Adoption of price controls and other cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures reforms may prevent or limit our ability to generate revenue, attain profitability or commercialize our product candidates.
Other healthcare laws and compliance requirements
We will also be subject to healthcare regulation and enforcement by the federal, state and foreign governments in which we will conduct our business once our products are approved. These fraud and abuse and transparency laws may impact, among other things, our financial arrangements and proposed sales, marketing and education programs.
The federal Anti-Kickback Statute, prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return
73
for, either the referral of an individual for, or the purchase, order, or recommendation of, an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Moreover, the federal civil and criminal false claims laws, including the civil False Claims Act, which can be enforced through “qui tam” whistleblower actions, prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent, or making a false statement or record material to payment of a false claim or avoiding, decreasing, or concealing an obligation to pay money to the federal government. Additionally, the government may assert that a claim including items and services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act.
In addition, the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
The Physician Payments Sunshine Act requires applicable manufacturers of covered drugs to disclose payments and other transfers of value provided to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals as well as physician ownership and investment interests. Beginning in 2022, applicable manufactures will also be required to report such information regarding its payments and other transfers of value to physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified nurse anesthetists and certified nurse-midwives during the previous year.
The majority of states also have statutes or regulations similar to the aforementioned federal anti-kickback and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. In addition, we may be subject to reporting requirements under state transparency laws, as well as state laws that require pharmaceutical companies to comply with the industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government that otherwise restricts certain payments that may be made to health care providers and entities. In addition, certain states and local jurisdictions require the registration of pharmaceutical sales representatives.
Because of the breadth of these laws and the narrowness of available statutory and regulatory exceptions, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If we or our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including significant administrative, civil and criminal penalties, damages, fines, imprisonment, disgorgement, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, exclusion of products from reimbursement under U.S. federal or state healthcare programs, and the curtailment or restructuring of our operations.
Government regulation outside of the United States
In addition to regulations in the United States, we will be subject to a variety of regulations in other jurisdictions governing, among other things, clinical studies and any commercial sales and distribution of our products.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical studies or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical study application much like the IND prior to the commencement of human clinical studies.
The requirements and process governing the conduct of clinical studies, product licensing, pricing and reimbursement vary from country to country. If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
74
Data privacy and security
Numerous state, federal and foreign laws, including consumer protection laws and regulations, govern the collection, dissemination, processing, use, access to, confidentiality and security of personal information, including health-related information. In the United States, numerous federal and state laws and regulations, including state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws and regulations (e.g., Section 5 of the FTC Act), govern the collection, use, disclosure, and protection of health-related and other personal information could apply to our operations or the operations of our partners. For example, HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and their respective implementing regulations, imposes privacy, security and breach notification obligations on certain health care providers, health plans, and health care clearinghouses, known as covered entities, as well as their business associates that perform certain services involving creating, receiving, maintaining or transmitting individually identifiable health information for or on behalf of such covered entities as well as their covered subcontractors. Entities that are found to be in violation of HIPAA as the result of a breach of unsecured protected health information, a complaint about privacy practices or an audit by HHS, may be subject to significant civil, criminal and administrative fines and penalties and/or additional reporting and oversight obligations if required to enter into a resolution agreement and corrective action plan with HHS to settle allegations of HIPAA non-compliance. Further, entities that knowingly obtain, use, or disclose individually identifiable health information maintained by a HIPAA covered entity in a manner that is not authorized or permitted by HIPAA may be subject to criminal penalties.
Even when HIPAA does not apply, according to the FTC, violating consumers’ privacy rights or failing to take appropriate steps to keep consumers’ personal information secure may constitute unfair acts or practices in or affecting commerce in violation of Section 5(a) of the FTC Act. The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Individually identifiable health information is considered sensitive data that merits stronger safeguards.
In addition, certain state and non-U.S. laws, such as the GDPR govern the privacy and security of health information in certain circumstances, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. Failure to comply with these laws, where applicable, can result in the imposition of significant civil and/or criminal penalties and private litigation. For example, California recently enacted legislation, the California Consumer Privacy Act, or CCPA, which went into effect January 1, 2020. The CCPA, among other things, creates new data privacy obligations for covered companies and provides new privacy rights to California residents, including the right to opt out of certain disclosures of their information. The CCPA also creates a private right of action with statutory damages for certain data breaches, thereby potentially increasing risks associated with a data breach. In Europe, the GDPR went into effect in May 2018 and introduces strict requirements for processing the personal data of individuals within the EEA. In addition, the GDPR increases the scrutiny of transfers of personal data from clinical trial sites located in the EEA to the United States and other jurisdictions that the European Commission does not recognize as having “adequate” data protection laws. Recent legal developments in Europe have created complexity and compliance uncertainty regarding certain transfers of personal data from the EEA. For example, on July 16, 2020, the Court of Justice of the European Union (“CJEU”) invalidated the EU-US Privacy Shield Framework (“Privacy Shield”) under which personal data could be transferred from the EEA.to United States entities who had self-certified under the Privacy Shield scheme. While the CJEU upheld the adequacy of the standard contractual clauses (a standard form of contract approved by the European Commission as an adequate personal data transfer mechanism, and potential alternative to the Privacy Shield), it made clear that reliance on them alone may not necessarily be sufficient in all circumstances. Use of the standard contractual clauses must now be assessed on a case-by-case basis taking into account the legal regime applicable in the destination country, in particular applicable surveillance laws and rights of individuals. Companies that must comply with the GDPR face increased compliance obligations and risk, including more robust regulatory enforcement of data protection requirements and potential fines for noncompliance of up to €20 million or 4% of the annual global revenues of the noncompliant company, whichever is greater. In addition, following Brexit and the end of the Transition Period, companies will have to comply with the GDPR and the GDPR as incorporated into United Kingdom national law, the latter regime having the ability to separately fine up to the greater of £17.5 million or 4% of global turnover. The relationship between the United Kingdom and the European Union in relation to certain aspects of data protection law remains unclear, for example around how data can lawfully be transferred between each jurisdiction, which exposes us to further compliance risk.
75
Employees and Human Capital Resources
As of December 31, 2020, we had 33 full-time employees in activities such as research and development, finance, clinical development, regulatory affairs, project management and administration. None of our employees is represented by a labor union or covered by a collective bargaining agreement. We consider our relationship with our employees to be good.
Our human capital objectives include, as applicable, identifying, recruiting, retaining, incentivizing and integrating our existing and additional employees. The principal purposes of our equity incentive plans are to attract, retain and motivate selected employees, consultants and directors through the granting of stock-based compensation awards.
In order to successfully implement our development and commercialization plans and strategies, and as we transition into operating as a public company, we expect to need additional managerial, operational, sales, marketing, financial and other personnel. Future growth would impose significant added responsibilities on members of management, including:
|
• |
identifying, recruiting, integrating, maintaining and motivating additional employees; |
|
• |
managing our internal development efforts effectively, including the clinical, FDA, EMA and other comparable foreign regulatory agencies’ review process for BT-11, NX-13 and any other future product candidates, while complying with any contractual obligations to contractors and other third parties we may have; and |
|
• |
improving our operational, financial and management controls, reporting systems and procedures. |
Available Information
Our internet website address is www.landosbiopharma.com. In addition to the information about us and our subsidiaries contained in this Annual Report, information about us can be found on our website. Our website and information included in or linked to our website are not part of this Annual Report.
Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, are available free of charge through our website as soon as reasonably practicable after they are electronically filed with or furnished to the Securities and Exchange Commission, or SEC. Additionally the SEC maintains an internet site that contains reports, proxy and information statements and other information. The address of the SEC's website is www.sec.gov.
The following information sets forth risk factors that could cause our actual results to differ materially from those contained in forward-looking statements we have made in this Annual Report on Form 10-K and those we may make from time to time. You should carefully consider the risks described below, in addition to the other information contained in this Annual Report on Form 10-K and our other public filings. Our business, financial condition or results of operations could be harmed by any of these risks. The risks and uncertainties described below are not the only ones we face. Additional risks not presently known to us or other factors not perceived by us to present significant risks to our business at this time also may impair our business operations.
Selected Risks Affecting Our Business
Our business is subject to a number of risks of which you should be aware before making a decision to invest in our common stock. These risks are more fully described in this “Risk Factors” section, including the following:
|
• |
We have incurred significant losses since our inception. We expect to incur losses over the next several years and may never achieve or maintain profitability. |
|
• |
We have a limited operating history, have not yet completed Phase 3 clinical trials and have no history of commercializing products, which may make it difficult for an investor to evaluate the success of our business to date and to assess our future viability. |
|
• |
We will need substantial additional funding to meet our financial obligations and to pursue our business objectives. If we are unable to raise capital when needed, we could be forced to curtail our planned operations and the pursuit of our growth strategy. |
76
|
• |
Our business and operations may be adversely affected by the evolving and ongoing COVID-19 global pandemic. |
|
• |
We currently have only two clinical-stage product candidates, BT-11 and NX-13. If we are unable to successfully develop, receive regulatory approval for and commercialize our product candidates for these or any other indications, or successfully develop any other product candidates, or experience significant delays in doing so, our business will be harmed. |
|
• |
The regulatory approval processes of the FDA and comparable foreign authorities are lengthy, time consuming and inherently unpredictable. If we are not able to obtain required regulatory approval for our product candidates, our business will be substantially harmed. |
|
• |
Success in preclinical studies or earlier clinical trials may not be indicative of results in future clinical trials. |
|
• |
We may not be successful in our efforts to increase our pipeline of product candidates, including by pursuing additional indications for our current product candidates or in-licensing or acquiring additional product candidates for other diseases. |
|
• |
We face substantial competition, which may result in a smaller than expected commercial opportunity and/or others discovering, developing or commercializing products before or more successfully than we do. |
|
• |
We rely on third parties to conduct a significant portion of our existing clinical trials and potential future clinical trials for product candidates, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials. |
|
• |
If we are unable to obtain or protect intellectual property rights related to any of our product candidates, we may not be able to compete effectively in our market. |
|
• |
An active trading market for our common stock may not develop. |
Risks related to our financial position and capital needs
We have incurred significant losses since our inception. We expect to incur losses over the next several years and may never achieve or maintain profitability.
We are a clinical-stage biopharmaceutical company with a limited operating history. Since our inception, we have incurred significant net losses, and we expect to continue to incur significant expenses and operating losses for the foreseeable future. Our net losses were $13.5 million and $30.1 million for the years ended December 31, 2019 and 2020, respectively. As of December 31, 2020, we had an accumulated deficit of $55.7 million. We have financed our operations with $70.0 million in gross proceeds raised in our private placements of convertible preferred stock and convertible promissory notes. On February 3, 2021, we completed our initial public offering (“IPO”) in which we issued and sold 6,250,000 shares of our common stock at a public offering price of $16 per share for net proceeds of $91.2 million, after deducting underwriters’ discounts and commissions.We have no products approved for commercialization and have never generated any revenue from product sales.
We have devoted substantially all of our financial resources and efforts to the development of our clinical and preclinical product candidates and our LANCE platform, including conducting preclinical studies and clinical trials. We expect to continue to incur significant expenses and operating losses over the next several years. We expect that it could be several years, if ever, before we have a commercialized product. Our net losses may fluctuate significantly from quarter to quarter and year to year. We anticipate that our expenses will increase substantially as we:
|
• |
conduct our ongoing and planned clinical trials of BT-11 and NX-13, as well as initiate and complete additional clinical trials; |
|
• |
pursue regulatory approval of BT-11 and NX-13 for the treatment of ulcerative colitis, or UC, and Crohn’s disease; |
|
• |
seek to discover and develop additional clinical and preclinical product candidates; |
|
• |
scale up our clinical and regulatory capabilities; |
77
|
• |
establish a commercialization infrastructure and scale up external manufacturing and distribution capabilities to commercialize any product candidates for which we may obtain regulatory approval, including BT-11 and NX-13; |
|
• |
adapt our regulatory compliance efforts to incorporate requirements applicable to marketed products; |
|
• |
maintain, expand and protect our intellectual property portfolio; |
|
• |
hire additional clinical, manufacturing and scientific personnel; |
|
• |
add operational, financial and management information systems and personnel, including personnel to support our product development and planned future commercialization efforts; and |
|
• |
incur additional legal, accounting and other expenses in operating as a public company. |
To become and remain profitable, we must succeed in developing and eventually commercializing product candidates that generate significant revenue. This will require us to be successful in a range of challenging activities, including completing preclinical testing and clinical trials of our product candidates, obtaining regulatory approval, and manufacturing, marketing and selling any product candidates for which we may obtain regulatory approval, as well as discovering and developing additional product candidates. We are only in the preliminary stages of most of these activities. We may never succeed in these activities and, even if we do, may never generate any revenue or revenue that is significant enough to achieve profitability.
Even if we achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would depress the value of our company and could impair our ability to raise capital, expand our business, maintain our development efforts, obtain product approvals, diversify our offerings or continue our operations. A decline in the value of our company could also cause you to lose all or part of your investment.
We have a limited operating history, have not yet completed Phase 3 clinical trials and have no history of commercializing products, which may make it difficult for an investor to evaluate the success of our business to date and to assess our future viability.
We commenced operations in 2017, and our operations to date have been largely focused on raising capital and developing our clinical and preclinical product candidates and our LANCE platform, including undertaking preclinical studies and conducting clinical trials. To date, we have not yet demonstrated our ability to successfully complete pivotal clinical trials, obtain regulatory approvals, manufacture a product on a commercial scale, or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful commercialization. Consequently, any predictions you make about our future success or viability may not be as accurate as they could be if we had a longer operating history or a history of successfully developing and commercializing products.
We may also need to transition from a company with a research focus to a company capable of supporting commercial activities. Our inability to adequately address these risks and difficulties or successfully make such a transition could adversely affect our business, financial condition, results of operations and growth prospects.
We may encounter unforeseen expenses, difficulties, complications, delays and other known or unknown factors in achieving our business objectives. We may also need to transition from a company with a research focus to a company capable of supporting commercial activities. Our inability to adequately address these risks and difficulties or successfully make such a transition could adversely affect our business, financial condition, results of operations and growth prospects.
We will need substantial additional funding to meet our financial obligations and to pursue our business objectives. If we are unable to raise capital when needed, we could be forced to curtail our planned operations and the pursuit of our growth strategy.
Our operations have consumed substantial amounts of cash since inception. Identifying potential product candidates and conducting preclinical testing and clinical trials is a time-consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain regulatory approval and achieve product sales. We expect to continue to incur significant expenses and operating losses over the next several years as we complete our ongoing clinical trials of our product candidates, initiate future clinical trials of our
78
product candidates, seek marketing approval for BT-11 and NX-13 for the treatment of UC and Crohn’s disease and advance any of our other product candidates we may develop or otherwise acquire. In addition, our product candidates, if approved, may not achieve commercial success. Our revenue, if any, will be derived from sales of products that we do not expect to be commercially available
for a number of years, if at all. If we obtain marketing approval for BT-11, NX-13 or any other product candidates that we develop or otherwise acquire, we expect to incur significant commercialization expenses related to product sales, marketing, distribution and manufacturing. We also expect an increase in our expenses associated with creating additional infrastructure to support operations as a public company.
As of December 31, 2020, we had cash, cash equivalents and marketable securities of $28.1 million. We believe that the net proceeds from our IPO, together with our existing cash and cash equivalents, will be sufficient to fund our operating expenses and capital requirements into 2023. This estimate is based on assumptions that may prove to be wrong, and we could use our available capital resources sooner than we expect. Changes may occur beyond our control that would cause us to consume our available capital before that time, including changes in and progress of our development activities, acquisitions of additional product candidates, and changes in regulation. Our future capital requirements will depend on many factors, including:
|
• |
the scope, progress, costs and results of our ongoing and planned clinical trials of BT-11 and NX-13, as well as the associated costs, including any unforeseen costs we may incur as a result or preclinical study or clinical trial delays due to the COVID-19 pandemic or other delays; |
|
• |
the scope, progress, costs and results of preclinical development, laboratory testing and clinical trials for any future product candidates we may decide to pursue; |
|
• |
the extent to which we develop, in-license or acquire other product candidates and technologies; |
|
• |
the costs and timing of process development and manufacturing scale-up activities associated with our product candidates and other programs we advance them through preclinical and clinical development; |
|
• |
the number and development requirements of other product candidates that we may pursue; |
|
• |
the costs, timing and outcome of regulatory review of our product candidates; |
|
• |
the costs and timing of future commercialization activities, including product manufacturing, marketing, sales and distribution, for any of our product candidates for which we receive marketing approval; |
|
• |
the revenue, if any, received from commercial sales of our product candidates for which we receive marketing approval; |
|
• |
our ability to establish collaborations to commercialize BT-11, NX-13 or any of our other product candidates outside the United States; and |
|
• |
the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending any intellectual property-related claims; |
We will require additional capital to commercialize BT-11 and NX-13. If we receive regulatory approval for either of these product candidates, we expect to incur significant commercialization expenses related to product manufacturing, sales, marketing and distribution, depending on where we choose to commercialize. Additional funds may not be available on a timely basis, on favorable terms, or at all, and such funds, if raised, may not be sufficient to enable us to continue to implement our long-term business strategy. Further, our ability to raise additional capital may be adversely impacted by potential worsening global economic conditions and the recent disruptions to and volatility in the credit and financial markets in the United States and worldwide resulting from the ongoing COVID-19 pandemic. If we are unable to raise sufficient additional capital, we could be forced to curtail our planned operations and the pursuit of our growth strategy.
Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
Until such time, if ever, as we can generate substantial revenue, we may finance our cash needs through a combination of equity offerings, government or private party grants, debt financings and license and collaboration agreements. We do not currently have any other committed external source of funds. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing and preferred equity financing, if available, may involve agreements that
79
include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends.
If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may be required to relinquish valuable rights to our technologies, future revenue streams or product candidates, grant licenses on terms that may not be favorable to us or commit to future payment streams. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves. Market volatility resulting from the COVID-19 pandemic or other factors may further adversely impact our ability to access capital as and when needed.
Risks related to the discovery, development and commercialization of our product candidates
Our business and operations may be adversely affected by the evolving and ongoing COVID-19 global pandemic.
Our business and operations may be adversely affected by the effects of the recent and evolving COVID-19 virus, which was declared by the World Health Organization as a global pandemic. The COVID-19 pandemic has resulted in travel and other restrictions in order to reduce the spread of the disease, including public health directives and orders in the United States and the European Union that, among other things and for various periods of time, directed individuals to shelter at their places of residence, directed businesses and governmental agencies to cease non-essential operations at physical locations, prohibited certain non-essential gatherings and events and ordered cessation of non-essential travel. Although our employees are no longer working remotely after the expiration of such orders in Virginia, future remote work policies and similar government orders or other restrictions on the conduct of business operations related to the COVID-19 pandemic may negatively impact productivity and may disrupt our ongoing research and development activities and our clinical programs and timelines, the magnitude of which will depend, in part, on the length and severity of the restrictions and other limitations on our ability to conduct our business in the ordinary course. Further, such orders also may impact the availability or cost of materials, which would disrupt our supply chain and manufacturing efforts and could affect our ability to conduct ongoing and planned clinical trials and preparatory activities.
Although our ongoing and planned clinical trials have not been impacted by the COVID-19 pandemic to date, we may experience related disruptions in the future that could severely impact our clinical trials, including:
|
• |
delays, difficulties or a suspension in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff; |
|
• |
risk that participants enrolled in our clinical trials will contract COVID-19 while the clinical trial is ongoing, which could impact the results of the clinical trial, including by increasing the number of observed adverse events; |
|
• |
interruptions in our ability to manufacture and deliver drug supply for trials; |
|
• |
diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials; |
|
• |
changes in local regulations as part of a response to the COVID-19 outbreak that may require us to change the ways in which our clinical trials are conducted, which may result in unexpected costs, or to discontinue the clinical trials altogether; |
|
• |
interruption of key clinical trial activities, such as clinical trial site monitoring, and the ability or willingness of subjects to travel to trial sites due to limitations on travel imposed or recommended by federal or state governments, employers and others; |
|
• |
limitations in employee resources that would otherwise be focused on the conduct of our clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people; |
|
• |
delays in necessary interactions with local regulators, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees; and |
|
• |
refusal of the United States Food and Drug Administration, or the FDA, to accept data from clinical trials in these affected geographies. |
The spread of COVID-19, which has caused a broad impact globally, may materially affect us economically. While the potential economic impact brought by, and the duration of, COVID-19 may be difficult to assess or predict, a widespread pandemic could result in significant disruption of global financial markets, reducing our ability to access
80
capital, which could in the future negatively affect our liquidity. In addition, a recession or market correction resulting from the spread of COVID-19 could materially affect our business and the value of our common stock.
The global COVID-19 pandemic continues to rapidly evolve. The extent to which the COVID-19 pandemic impacts our business and operations, including our clinical development and regulatory efforts, will depend on future developments that are highly uncertain and cannot be predicted with confidence at the time of this Annual Report, such as the ultimate geographic spread of the disease, the duration of the outbreak, the duration and effect of business disruptions and the short-term effects and ultimate effectiveness of the travel restrictions, quarantines, social distancing requirements and business closures in the United States and other countries to contain and treat the disease. Accordingly, we do not yet know the full extent of potential delays or impacts on our business, our clinical and regulatory activities, healthcare systems or the global economy as a whole. However, these impacts could adversely affect our business, financial condition, results of operations and growth prospects.
In addition, to the extent the ongoing COVID-19 pandemic adversely affects our business and results of operations, it may also have the effect of heightening many of the other risks and uncertainties described in this “Risk factors” section.
We currently have only two clinical-stage product candidates, BT-11 and NX-13. If we are unable to successfully develop, receive regulatory approval for and commercialize such product candidates for these or any other indications, or successfully develop any other product candidates, or experience significant delays in doing so, our business will be harmed.
We currently have no products that are approved for commercial sale. We currently have only two clinical-stage product candidates, BT-11 and NX-13. To date, we have not yet conducted any pivotal clinical trials. We have not completed the development of any product candidates, and we may never be able to develop marketable products.
We have invested substantially all of our efforts and financial resources in the development of our clinical and preclinical product candidates and our LANCE platform. Our ability to generate revenue from our product candidates, which we do not expect will occur for several years, if ever, will depend heavily on the successful development, regulatory approval and eventual commercialization of our product candidates. The success of BT-11, NX-13 or any other product candidates that we develop or otherwise may acquire will depend on several factors, including:
|
• |
timely and successful completion of preclinical studies and clinical trials; |
|
• |
sufficiency of our financial and other resources to complete the necessary preclinical studies and clinical trials; |
|
• |
successful patient enrollment and completion of clinical trials; |
|
• |
successful development of, or making arrangements with third-party manufacturers for, our commercial manufacturing processes for any of our product candidates that receive regulatory approval; |
|
• |
timely receipt marketing approvals from applicable regulatory authorities; |
|
• |
launching commercial sales of products, if approved; |
|
• |
acceptance of our products, if approved, by patients, the medical community and third-party payors, for their approved indications; |
|
• |
the prevalence and severity of adverse events experienced with BT-11, NX-13 or any other product candidates; |
|
• |
the availability, perceived advantages, cost, safety and efficacy of alternative therapies for any product candidate, and any indications for such product candidate, that we develop, specifically alternative treatments for UC or Crohn’s disease; |
|
• |
our ability to produce BT-11, NX-13 or any other product candidates we develop on a commercial scale; |
|
• |
obtaining and maintaining patent, trademark and trade secret protection and regulatory exclusivity for our product candidates and otherwise protecting our rights in our intellectual property portfolio; |
|
• |
maintaining compliance with regulatory requirements, including current good manufacturing practices, or cGMPs, and complying effectively with other procedures; |
|
• |
maintaining a continued acceptable safety, tolerability and efficacy profile of the products following approval; |
81
|
• |
obtaining and maintaining patent protection, trade secret protection and regulatory exclusivity, both in the United States and internationally; |
|
• |
the protection of our rights in our intellectual property portfolio; |
|
• |
the successful launch of commercial sales following any marketing approval; |
|
• |
a continued acceptable safety profile following any marketing approval; |
|
• |
commercial acceptance by patients, the medical community and third-party payors; and |
|
• |
our ability to compete with other therapies. |
We do not have complete control over many of these factors, including certain aspects of clinical development and the regulatory submission process, potential threats to our intellectual property rights and the manufacturing, marketing, distribution and sales efforts of any future collaborator. If we are not successful with respect to one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully commercialize the product candidates we develop, which would materially harm our business. If we do not receive marketing approvals for BT-11, NX-13 or any other product candidate we develop, we may not be able to continue our operations.
The regulatory approval processes of the FDA and comparable foreign authorities are lengthy, time consuming and inherently unpredictable. If we are not able to obtain required regulatory approval for our product candidates, our business will be substantially harmed.
The time required to obtain approval or other marketing authorizations by the FDA and comparable foreign authorities is unpredictable, and it typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, and the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. BT-11 and NX-13 are currently our only clinical-stage product candidates. We have not obtained regulatory approval for any product candidate, and it is possible that we may never obtain regulatory approval for BT-11, NX-13 or any product candidates we may seek to develop in the future. Neither we nor any current or future collaborator is permitted to market any drug product candidates in the United States until we receive regulatory approval of a new drug application, or NDA, from the FDA. To date, we have only had limited discussions with the European Medicines Agency, or EMA, and other comparable foreign authorities regarding regulatory approval for BT-11, NX-13 or any other product candidate outside of the United States.
Prior to obtaining approval to commercialize any drug product candidate in the United States or abroad, we must demonstrate with substantial evidence from well-controlled clinical trials, and to the satisfaction of the FDA or foreign regulatory agencies, that such product candidates are safe, pure and effective for their intended uses. Results from preclinical studies and clinical trials can be interpreted in different ways. Even if we believe the preclinical or clinical data for our product candidates are promising, such data may not be sufficient to support approval by the FDA and other regulatory authorities. The FDA may also require us to conduct additional preclinical studies or clinical trials for our product candidates either prior to or after approval, or it may object to elements of our clinical development programs.
Of the large number of products in development, only a small percentage successfully complete the FDA or foreign regulatory approval processes and are commercialized. The lengthy approval and marketing authorization process as well as the unpredictability of future clinical trial results may result in our failing to obtain regulatory approval and marketing authorization to market our product candidates, which would significantly harm our business, financial condition, results of operations and prospects.
We have invested a significant portion of our time and financial resources in the development of our clinical and preclinical product candidates, including BT-11 and NX-13. Our business is dependent on our ability to successfully complete preclinical and clinical development of, obtain regulatory approval for, and, if approved, successfully commercialize BT-11, NX-13 and any future product candidates in a timely manner.
Even if we eventually complete clinical testing and receive approval of a NDA or foreign marketing application for BT-11, NX-13 or any future product candidates, the FDA or the applicable foreign regulatory agency may grant approval or other marketing authorization contingent on the performance of costly additional clinical trials, including post-marketing clinical trials. The FDA or the applicable foreign regulatory agency also may approve or authorize for marketing a product candidate for a more limited indication or patient population than we originally request, and the FDA or applicable foreign regulatory agency may not approve or authorize the labeling that we believe is necessary or desirable for the successful commercialization of a product candidate. Any delay in obtaining, or inability to obtain, applicable regulatory approval or other marketing authorization would delay or
82
prevent commercialization of that product candidate and would materially adversely impact our business and prospects.
In addition, the FDA and other regulatory authorities may change their policies, issue additional regulations or revise existing regulations, or take other actions, which may prevent or delay approval of our future products under development on a timely basis. Such policy or regulatory changes could impose additional requirements upon us that could delay our ability to obtain approvals, increase the costs of compliance or restrict our ability to maintain any marketing authorizations we may have obtained.
Clinical product development involves a lengthy and expensive process, with an uncertain outcome. We may incur additional costs and experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
In order to obtain FDA approval to market a new drug product we must demonstrate proof of safety, purity and efficacy in humans. The risk of failure for product candidates is high. It is impossible to predict when or if any of our product candidates will prove effective or safe in humans or will receive regulatory approval. Before obtaining marketing approval from regulatory authorities for the sale of any product candidate, we must complete preclinical development and then conduct extensive clinical trials to demonstrate the safety, purity, potency, and efficacy of our product candidates in humans. Clinical testing is expensive, difficult to design and implement, can take many years to complete and is inherently uncertain as to outcome. A failure of one or more clinical trials can occur at any stage of testing or at any time during the trial process. The outcome of preclinical testing and early clinical trials may not be predictive of the results of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval of their products.
We have not completed all clinical trials required for the approval of any of our product candidates. We cannot assure you that any clinical trial that we are conducting, or may conduct in the future, will demonstrate consistent or adequate efficacy and safety to obtain regulatory approval to market our product candidates.
We may incur additional costs and experience delays in ongoing clinical trials for our product candidates, and we do not know whether future clinical trials, if any, will begin on time, need to be redesigned, enroll an adequate number of patients on time or be completed on schedule, if at all. We may experience numerous unforeseen events during or as a result of clinical trials that could delay or prevent our ability to receive marketing approval or commercialize our product candidates, including:
|
• |
regulators or institutional review boards may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; |
|
• |
we may experience delays in reaching, or fail to reach, agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites or prospective contract research organizations, or CROs, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
|
• |
clinical trials of our product candidates may produce negative or inconclusive results, including failure to demonstrate statistical significance, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs; |
|
• |
we may fail to demonstrate statistical significance in early stage or Phase II clinical trials of our product candidates, which may impact the timing and design of late stage clinical trials for such product candidates; |
|
• |
the number of patients required for clinical trials of our product candidates may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials or fail to return for post-treatment follow-up at a higher rate than we anticipate; |
|
• |
our product candidates may have undesirable side effects or other unexpected characteristics, causing us or our investigators, regulators or institutional review boards to suspend or terminate the trials; |
|
• |
our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all; |
83
|
• |
regulators or institutional review boards may require that we or our investigators suspend or terminate clinical development for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks; |
|
• |
the cost of clinical trials of our product candidates may be greater than we anticipate; and |
|
• |
the supply or quality of our product candidates or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate. |
If we experience delays in the completion of, or termination of, any clinical trial of our product candidates, the commercial prospects of our product candidates will be harmed, and our ability to generate product revenues from any of these product candidates will be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues.
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete clinical trials of our product candidates or other testing, if the results of these trials or tests are not favorable or if there are safety concerns, we may:
|
• |
be delayed in obtaining marketing approval for our product candidates; |
|
• |
not obtain marketing approval at all; |
|
• |
obtain approval for indications or patient populations that are not as broad as intended or desired; |
|
• |
obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; |
|
• |
be subject to additional post-marketing testing requirements; or |
|
• |
have the product removed from the market after obtaining marketing approval. |
Success in preclinical studies or earlier clinical trials may not be indicative of results in future clinical trials.
Success in preclinical testing and early clinical trials does not ensure that later clinical trials will generate the same results or otherwise provide adequate data to demonstrate the efficacy and safety of a product candidate. Preclinical tests and Phase 1 and Phase 2 clinical trials are primarily designed to test safety, to study pharmacokinetics and pharmacodynamics and to understand the side effects of product candidates at various doses and schedules. Success in preclinical or animal studies and early clinical trials does not ensure that later large-scale efficacy trials will be successful nor does it predict final results. Our product candidates may fail to show the desired safety and efficacy in clinical development despite positive results in preclinical studies or having successfully advanced through initial clinical trials, particularly because we are targeting novel pathways that have not yet been tested in later-stage clinical trials.
Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in preclinical testing and earlier-stage clinical trials. Data obtained from preclinical and clinical activities are subject to varying interpretations, which may delay, limit or prevent regulatory approval. In addition, we may experience regulatory delays or rejections as a result of many factors, including changes in regulatory policy during the period of our product candidate development. Any such delays could negatively impact our business, financial condition, results of operations and prospects.
We may seek orphan drug designation for some of our product candidates and we may be unsuccessful, or may be unable to maintain the benefits associated with orphan drug designation, including the potential for market exclusivity, for product candidates for which we obtain orphan drug designation.
Regulatory authorities in some jurisdictions, including the United States, may designate drugs or biologics intended to treat relatively small patient populations as orphan drug products. Under the Orphan Drug Act, the FDA may designate a drug or biologic as an orphan drug if it is intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States.
84
If a drug or biologic with an orphan drug designation subsequently receives the first marketing approval for the indication for which it has such designation, the drug or biologic is entitled to a period of marketing exclusivity, which precludes the FDA from approving another marketing application for the same drug and indication for that time period, except in limited circumstances. If our competitors are able to obtain orphan drug exclusivity prior to us, for products that constitute the “same drug” and treat the same indications as our product candidates, we may not be able to have competing products approved by the applicable regulatory authority for a significant period of time. The applicable period is seven years in the United States.
We intend to seek orphan designation for a modified formulation of BT-11 for the treatment of eosinophilic esophagitis. However, we may be unsuccessful in obtaining orphan drug designation for this or other product candidates, and may be unable to maintain the benefits associated with orphan drug designation. Even if we obtain orphan drug exclusivity for any of our product candidates, that exclusivity may not effectively protect those product candidates from competition because different drugs can be approved for the same condition, and orphan drug exclusivity does not prevent the FDA from approving the same or a different drug in another indication. Even after an orphan drug is granted orphan exclusivity and approved, the FDA can subsequently approve a later application for the same drug for the same condition before the expiration of the seven-year exclusivity period if the FDA concludes that the later drug is clinically superior in that it is shown to be safer in a substantial portion of the target populations, more effective or makes a major contribution to patient care. In addition, a designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. Moreover, orphan-drug-exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if we are unable to manufacture sufficient quantities of the product to meet the needs of patients with the rare disease or condition. Orphan drug designation neither shortens the development time or regulatory review time of a drug nor gives the drug any advantage in the regulatory review or approval process.
Interim “top-line” and preliminary results from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publish interim top-line or preliminary results from our preclinical studies and clinical trials, which are based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study or trial. Interim results from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Preliminary or top-line results also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, interim and preliminary data should be viewed with caution until the final data are available. Differences between preliminary or interim data and final data could significantly harm our business prospects and may cause the trading price of our common stock to fluctuate significantly.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates,
calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and investors or others may not agree with what we determine is material or otherwise appropriate information to include in our disclosure.
Our clinical trials may fail to demonstrate the safety and efficacy of our product candidates, or serious adverse or unacceptable side effects may be identified during the development of our product candidates, which could prevent or delay regulatory approval and commercialization, increase our costs or necessitate the abandonment or limitation of the development of some of our product candidates.
Before obtaining regulatory approvals for the commercial sale of our product candidates, we must demonstrate through lengthy, complex and expensive preclinical testing and clinical trials that our product candidates are safe, pure and effective for use in each target indication, and failures can occur at any stage of testing. Clinical trials often fail to demonstrate safety or efficacy of the product candidate studied for the target indication.
If our product candidates are associated with side effects in clinical trials or have characteristics that are unexpected, we may need to abandon their development or limit development to more narrow uses in which the side effects or
85
other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. The FDA or an institutional review board may also require that we suspend, discontinue, or limit our clinical trials based on safety information, or that we conduct additional animal or human studies regarding the safety and efficacy of our product candidates which we have not planned or anticipated. Such findings could further result in regulatory authorities failing to provide marketing authorization for our product candidates or limiting the scope of the approved indication, if approved. Many product candidates that initially showed promise in early stage testing have later been found to cause side effects that prevented further development of the product candidate.
Additionally, if one or more of our product candidates receives marketing approval, and we or others identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
|
• |
regulatory authorities may withdraw approvals of such product; |
|
• |
regulatory authorities may require additional warnings on the labels; |
|
• |
we may be required to create a medication guide outlining the risks of such side effects for distribution to patients; |
|
• |
we could be sued and held liable for harm caused to patients; |
|
• |
we may not be able to achieve or maintain third-party payor coverage and adequate reimbursement; and |
|
• |
our reputation and physician or patient acceptance of our products may suffer. |
There can be no assurance that we will resolve any issues related to any product-related adverse events to the satisfaction of the FDA or foreign regulatory agency in a timely manner or at all. Moreover, any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, results of operations and prospects.
As an organization, we have never conducted pivotal clinical trials, and we may be unable to do so for any product candidates we may develop.
We will need to successfully complete pivotal clinical trials in order to obtain the approval of the FDA, EMA or other regulatory agencies to market BT-11, NX-13 or any future product candidate. Carrying out pivotal clinical trials is a complicated process. As an organization, we have not previously conducted any later stage or pivotal clinical trials. In order to do so, we will need to expand our clinical development and regulatory capabilities, and we may be unable to recruit and train qualified personnel. We also expect to continue to rely on third parties to conduct our pivotal clinical trials. See “—Risks related to our dependence on third parties—We will rely on third parties to conduct our future clinical trials for product candidates, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials.” Consequently, we may be unable to successfully and efficiently execute and complete necessary clinical trials in a way that leads to NDA submission and approval of BT-11, NX-13 or future product candidates. We may require more time and incur greater costs than our competitors and may not succeed in obtaining regulatory approvals of product candidates that we develop. Failure to commence or complete, or delays in, our planned clinical trials, could prevent us from or delay us in commercializing our product candidates.
If we experience delays or difficulties in the enrollment and/or maintenance of patients in clinical trials, our receipt of necessary regulatory approvals could be delayed or prevented.
Successful and timely completion of clinical trials will require that we enroll a sufficient number of patients. Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors, including the size and nature of the patient population and competition for patients with other trials. For example, we are aware of multiple clinical trials for the treatment of UC and Crohn’s disease being conducted by competitors that may make it difficult for us to enroll sufficient patients. Trials may be subject to delays as a result of patient enrollment taking longer than anticipated or patient withdrawal. We may not be able to initiate or continue clinical trials for our product candidates if we are unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or foreign regulatory authorities. We cannot predict how successful we will be at enrolling subjects in future clinical trials. Subject enrollment is affected by other factors including:
|
• |
the eligibility criteria for the trial in question; |
|
• |
the size of the patient population and process for identifying patients; |
|
• |
the perceived risks and benefits of the product candidate in the trial; |
86
|
• |
the availability of competing commercially available therapies and other competing drug candidates’ clinical trials; |
|
• |
the willingness of patients to be enrolled in our clinical trials; |
|
• |
the efforts to facilitate timely enrollment in clinical trials; |
|
• |
potential disruptions caused by the COVID-19 pandemic, including difficulties in initiating clinical sites, enrolling and retaining participants, diversion of healthcare resources away from clinical trials, travel or quarantine policies that may be implemented, and other factors; |
|
• |
the patient referral practices of physicians; |
|
• |
the ability to monitor patients adequately during and after treatment; and |
|
• |
the proximity and availability of clinical trial sites for prospective patients. |
Our inability to enroll a sufficient number of patients for clinical trials would result in significant delays and could require us to abandon one or more clinical trials altogether. Enrollment delays in these clinical trials may result in increased development costs for our product candidates, which would cause the value of our company to decline and limit our ability to obtain additional financing. Furthermore, we rely on and expect to continue to rely on CROs and clinical trial sites to ensure the proper and timely conduct of our clinical trials and we will have limited influence over their performance.
Furthermore, even if we are able to enroll a sufficient number of patients for our clinical trials, we may have difficulty maintaining enrollment of such patients in our clinical trials.
Changes in methods of product candidate manufacturing or formulation may result in additional costs or delay.
As product candidates proceed through preclinical studies to late-stage clinical trials towards potential approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods and formulation, are altered along the way in an effort to optimize processes and product characteristics. Such changes carry the risk that they will not achieve these intended objectives. Any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials conducted with the materials manufactured using altered processes. Such changes may also require additional testing, FDA notification or FDA approval. This could delay completion of clinical trials, require the conduct of bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of our product candidates and jeopardize our ability to commence sales and generate revenue.
We may not be successful in our efforts to increase our pipeline of product candidates, including through our LANCE platform, by pursuing additional indications for our current product candidates or by in-licensing or acquiring additional product candidates for other diseases.
A key element of our strategy is to build and expand our pipeline of product candidates. We have developed the LANCE platform to enable the identification, testing, design and development of new product candidates. We cannot assure you that our LANCE platform will work, nor that any of these potential targets or other aspects of our platform will yield product candidates that could enter clinical development and, ultimately, be commercially valuable. Although we expect to continue to enhance the capabilities of our LANCE platform by developing and integrating existing and new research technologies, we may not be successful in any of our enhancement and development efforts. If our enhancement or development efforts are unsuccessful, we may not be able to advance our drug discovery capabilities as quickly as we expect or identify as many potential drug candidates as we desire.
In addition, we may in-license or acquire additional product candidates for other diseases. We may not be able to identify or develop product candidates that are safe, tolerable and effective. Even if we are successful in continuing to build our pipeline, the potential product candidates that we identify, in-license or acquire may not be suitable for clinical development, including as a result of being shown to have harmful side effects or other characteristics that indicate that they are unlikely to be products that will receive marketing approval and achieve market acceptance.
87
We may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and management resources, we focus on development programs and product candidates that we identify for specific indications. As such, we are currently primarily focused on the development of BT-11 and NX-13 for the treatment of UC and Crohn’s disease. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications for BT-11 and NX-13 that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future development programs and product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate.
We intend to explore the use of BT-11 and NX-13, and potentially other product candidates, in combination with other therapies, which exposes us to additional risks.
We intend to explore the use of BT-11 and NX-13 and likely other future product candidates in combination with one or more other approved or unapproved therapies to treat UC and CD.
Even if any product candidate we develop were to receive marketing approval or be commercialized for use in combination with other existing therapies, we would continue to be subject to the risks that the FDA, EMA or comparable foreign regulatory authorities outside of the United States could revoke approval of the therapy used in combination with our product or that safety, efficacy, manufacturing or supply issues could arise with any of those existing therapies. If the therapies we use in combination with our product candidates are replaced as the standard of care for the indications we choose for any of our product candidates, the FDA, EMA or comparable foreign regulatory authorities may require us to conduct additional clinical trials. The occurrence of any of these risks could result in our own products, if approved, being removed from the market or being less successful commercially.
We also may choose to evaluate BT-11 and NX-13 or any other future product candidates in combination with one or more therapies that have not yet been approved for marketing by the FDA, EMA or comparable foreign regulatory authorities. We will not be able to market and sell BT-11 or NX-13 or any product candidate we develop in combination with an unapproved therapy for a combination indication if that unapproved therapy does not ultimately obtain marketing approval either alone or in combination with our product. In addition, unapproved therapies face the same risks described with respect to our product candidates currently in development and clinical trials, including the potential for serious adverse effects, delay in their clinical trials and lack of FDA approval.
If the FDA, EMA or comparable foreign regulatory authorities do not approve these other drugs or revoke their approval of, or if safety, efficacy, quality, manufacturing or supply issues arise with, the drugs we choose to evaluate in combination with our product candidate we develop, we may be unable to obtain approval of or market such combination therapy.
The United Kingdom’s withdrawal from the European Union may have a negative effect on global economic conditions, financial markets and our business.
Following the result of a referendum in 2016, the United Kingdom left the European Union on January 31, 2020, commonly referred to as Brexit.
88
Since a significant proportion of the regulatory framework in the United Kingdom applicable to our business and our product candidates is derived from European Union directives and regulations, Brexit, could materially impact the regulatory regime with respect to the development, manufacture, importation, approval and commercialization of our product candidates in the United Kingdom or the European Union. For example, as a result of the uncertainty surrounding Brexit, the EMA relocated to Amsterdam from London. The United Kingdom will no longer be covered by the centralized procedures for obtaining European Union-wide marketing and manufacturing authorizations from the EMA and a separate process for authorization of drug products will be required in the United Kingdom, the potential process for which is currently unclear. Any delay in obtaining, or an inability to obtain, any marketing approvals, as a result of Brexit or otherwise, would prevent us from commercializing our product candidates in the United Kingdom or the European Union and limit our ability to generate revenue and achieve and sustain profitability. In addition, we may be required to pay taxes or duties or be subjected to other hurdles in connection with the importation of our product candidates into the European Union, or we may incur expenses in establishing a manufacturing facility in the European Union in order to circumvent such hurdles. If any of these outcomes occur, we may be forced to restrict or delay efforts to seek regulatory approval in the United Kingdom or the European Union for our product candidates, or incur significant additional expenses to operate our business, which could significantly and materially harm or delay our ability to generate revenues or achieve profitability of our business. Any further changes in international trade, tariff and import/export regulations as a result of Brexit or otherwise may impose unexpected duty costs or other non-tariff barriers on us. These developments, or the perception that any of them could occur, may significantly reduce global trade and, in particular, trade between the impacted nations and the United Kingdom. It is also possible that Brexit may negatively affect our ability to attract and retain employees, particularly those from the European Union.
Risks related to the commercialization of our product candidates
Even if any of our product candidates receive marketing approval, they may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success.
If any of our product candidates receive marketing approval, they may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community. If our product candidates do not achieve an adequate level of acceptance, we may not generate significant revenue and we may not become profitable. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on a number of factors, including:
|
• |
the efficacy, safety and potential advantages compared to alternative treatments; |
|
• |
the timing of market introduction of the product candidate as well as competitive products; |
|
• |
our ability to offer our products for sale at competitive prices; |
|
• |
the convenience and ease of administration compared to alternative treatments; |
|
• |
product labeling or product insert requirements of the FDA or foreign regulatory authorities, including any limitations or warnings contained in a product’s approved labeling, including any black box warning; |
|
• |
the availability of the approved product candidate for use as a combination therapy; |
|
• |
the willingness of the target patient population to try new treatments and of physicians to prescribe these treatments; |
|
• |
our ability to hire and retain a sales force in the United States; |
|
• |
the strength of marketing and distribution support; |
|
• |
the availability of third-party coverage and adequate reimbursement for BT-11, NX-13 and any other product candidates, once approved; |
|
• |
the prevalence and severity of any side effects; and |
|
• |
any restrictions on the use of our products together with other medications. |
89
If we are unable to establish sales, marketing and distribution capabilities for BT-11, NX-13 or any other product candidate that may receive regulatory approval, we may not be successful in commercializing those product candidates if and when they are approved.
We do not have sales or marketing infrastructure. To achieve commercial success for BT-11, NX-13 or any other product candidate for which we may obtain marketing approval, we will need to establish a sales and marketing organization. In the future, we expect to build a focused sales and marketing infrastructure to market some of our product candidates in the United States, if and when they are approved. There are risks involved with establishing our own sales, marketing and distribution capabilities. For example, recruiting and training a sales force is expensive and time consuming and could delay any product launch. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
Factors that may inhibit our efforts to market our products on our own include:
|
• |
our inability to recruit, train and retain adequate numbers of effective sales and marketing personnel; |
|
• |
the inability of sales personnel to obtain access to physicians in order to educate physicians about our product candidates, once approved; |
|
• |
the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and |
|
• |
unforeseen costs and expenses associated with creating an independent sales and marketing organization. |
If we are unable to establish our own sales, marketing and distribution capabilities and are forced to enter into arrangements with, and rely on, third parties to perform these services, our revenue and our profitability, if any, are likely to be lower than if we had developed such capabilities ourselves. In addition, we may not be successful in entering into arrangements with third parties to sell, market and distribute our product candidates or may be unable to do so on terms that are favorable to us. We likely will have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively. If we do not establish sales, marketing and distribution capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidates.
We face substantial competition, which may result in a smaller than expected commercial opportunity and/or others discovering, developing or commercializing products before or more successfully than we do.
The biotechnology and pharmaceutical industries, and particularly the market for the treatment of autoimmune diseases, are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary and novel products and product candidates. We face competition with respect to our current product candidates, and will face competition with respect to any product candidates that we may seek to develop or commercialize in the future, from many different sources, including major pharmaceutical and specialty pharmaceutical companies, compounding facilities, academic institutions and governmental agencies and public and private research institutions.
We are aware of several other products and product candidates as potential treatments for UC and Crohn’s disease that would compete with BT-11 and NX-13, if approved. In particular, we expect to compete against companies that produce biologic drugs that currently dominate the UC and Crohn’s disease market, as well as companies that produce the aminosalicylates, steroids and immunosuppressants that are currently used to treat patients with mild to moderate disease.
In addition, our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer or more effective, have fewer or less severe side effects, are more convenient or are less expensive than BT-11, NX-13 or any other product that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for our product, which could result in our competitors establishing a strong market position before we are able to enter the market.
90
Many of the companies against which we are competing, or against which we may compete in the future, have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or that may be necessary for, our programs.
The success of BT-11, NX-13 or any future product candidate, will depend significantly on coverage and adequate reimbursement or the willingness of patients to pay for these products.
We believe our success depends on obtaining and maintaining coverage and adequate reimbursement for BT-11 and NX-13 for the treatment of UC and Crohn’s disease, and the extent to which patients will be willing to pay out-of-pocket for such products, in the absence of reimbursement for all or part of the cost. Additionally, in the United States, there is no uniform policy of coverage and reimbursement among third-party payors. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own coverage and reimbursement policies. However, decisions regarding the extent of coverage and amount of reimbursement to be provided are made on a payor-by-payor basis. One payor’s determination to provide coverage for a drug product does not assure that other payors will also provide coverage, and adequate reimbursement.
Third-party payors determine which products they will cover and establish reimbursement levels. Even if a third-party payor covers a particular product, the resulting reimbursement payment rates may not be adequate. .Reimbursement by a third-party payor may depend upon a number of factors, including the third-party payor’s determination that a product is safe, effective and medically necessary; appropriate for the specific patient; cost-effective; supported by peer-reviewed medical journals; included in clinical practice guidelines; and neither cosmetic, experimental, nor investigational.
Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. Further, such payors are increasingly challenging the price, examining the medical necessity and reviewing the cost effectiveness of medical product candidates. There may be especially significant delays in obtaining coverage and reimbursement for newly approved drugs. Third-party payors may limit coverage to specific product candidates on an approved list, known as a formulary, which might not include all FDA-approved drugs for a particular indication. We may need to conduct expensive pharmaco-economic studies to demonstrate the medical necessity and cost effectiveness of our products. Nonetheless, our product candidates may not be considered medically necessary or cost effective. We cannot be sure that coverage and reimbursement will be available for any product that we commercialize and, if reimbursement is available, what the level of reimbursement will be.
Foreign governments also have their own healthcare reimbursement systems, which vary significantly by country and region, and we cannot be sure that coverage and adequate reimbursement will be made available with respect to the treatments in which our products are used under any foreign reimbursement system.
There can be no assurance that BT-11, NX-13 or any other product candidate, if approved for sale in the United States or in other countries, will be considered medically reasonable and necessary, that it will be considered cost-effective by third-party payors, that coverage or an adequate level of reimbursement will be available or that reimbursement policies and practices in the United States and in foreign countries where our products are sold will not adversely affect our ability to sell our product candidates profitably, if they are approved for sale.
The market for BT-11, NX-13 or any other product candidates may be smaller than we expect.
Our estimates of the potential market opportunity for BT-11, NX-13 or any other product candidates include several key assumptions based on our industry knowledge, industry publications and third-party research reports. These assumptions include the number of patients who have the autoimmune diseases we intend to target, as well as the estimated reimbursement levels for each product candidate if approved. However, there can be no assurance that any of these assumptions are, or will remain, accurate. Further, new studies may change the estimated incidence or prevalence of these diseases, and the potentially addressable patient population for our product candidates may not ultimately be amenable to treatment with our product candidates. If the actual market for BT-11, NX-13 or for any other product candidates we may develop is smaller than we expect, our revenues, if any, may be limited and it may be more difficult for us to achieve or maintain profitability.
91
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials and will face an even greater risk if we commercially sell any products that we may develop. If we cannot successfully defend ourselves against claims that our product candidates or drugs caused injuries, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
|
• |
decreased demand for any product candidates or drugs that we may develop; |
|
• |
injury to our reputation and significant negative media attention; |
|
• |
withdrawal of clinical trial participants; |
|
• |
significant costs to defend the related litigation; |
|
• |
substantial monetary awards paid to trial participants or patients; |
|
• |
loss of revenue; |
|
• |
reduced resources of our management to pursue our business strategy; and |
|
• |
the inability to commercialize any products that we may develop. |
Although we maintain product liability insurance coverage, such insurance may not be adequate to cover all liabilities that we may incur. We may need to increase our insurance coverage as we expand our clinical trials or if we commence commercialization of our product candidates. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise.
Risks related to our dependence on third parties
We rely on third parties to conduct a significant portion of our existing clinical trials and potential future clinical trials for product candidates, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials.
To date, we have generally engaged CROs to conduct our ongoing and completed clinical trials of BT-11 and NX-13. We expect to engage CROs for future clinical trials for BT-11, NX-13 or other product candidates that we may progress to clinical development. We expect to continue to rely on third parties, including clinical data management organizations, medical institutions and clinical investigators, to conduct those clinical trials. Any of these third parties may terminate their engagements with us, some in the event of an uncured material breach and some at any time for convenience. If any of our relationships with these third parties terminate, we may not be able to timely enter into arrangements with alternative third parties or to do so on commercially reasonable terms, if at all. Switching or adding CROs involves substantial cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can materially impact our ability to meet our desired clinical development timelines. Though we intend to carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects. Further, the performance of our CROs may also be interrupted by the ongoing COVID-19 pandemic, including due to travel or quarantine policies, heightened exposure of CRO staff who are healthcare providers to COVID-19 or prioritization of resources toward the pandemic.
In addition, any third parties conducting our clinical trials will not be our employees, and except for remedies available to us under our agreements with such third parties, we cannot control whether or not they devote sufficient time and resources to our clinical programs. If these third parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. Consequently, our results of operations and the commercial prospects for our product candidates would be harmed, our costs could increase substantially and our ability to generate revenue could be delayed significantly.
92
We rely on these parties for execution of our preclinical studies and clinical trials, and generally do not control their activities. Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, we will remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with standards, commonly referred to as good clinical practices, or GCPs, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, ClinicalTrials.gov, within specified timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions. If we or any of our CROs or other third parties, including trial sites, fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, EMA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials complies with GCP regulations. In addition, our clinical trials must be conducted with product produced under cGMP conditions. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
In addition, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and receive compensation in connection with such services. Under certain circumstances, we may be required to report some of these relationships to the FDA. The FDA may conclude that a financial relationship between us and a principal investigator has created a conflict of interest or otherwise affected interpretation of the trial. The FDA may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA and may ultimately lead to the denial of marketing approval of BT-11, NX-13 or any other product candidates.
We also expect to rely on other third parties to store and distribute product supplies for our clinical trials. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our product candidates or commercialization of our products, producing additional losses and depriving us of potential revenue.
We contract with third parties for the manufacture of BT-11 and NX-13 for clinical drug supply and expect to continue to do so for commercialization if approved. This reliance on third parties increases the risk that we will not have sufficient quantities of our product candidates or such quantities at an acceptable cost, which could delay, prevent or impair our development or commercialization efforts.
We do not have any cGMP manufacturing facilities. We currently rely, and expect to continue to rely, on third parties for the cGMP manufacture of BT-11, NX-13 and any other product candidates that we may pursue, for clinical development. Any significant delay, including any delays as a result of the COVID-19 pandemic, in the supply of a product candidate or raw material components for an ongoing clinical trial due to the need to replace a third-party CMO could considerably delay the completion of our clinical trials.
We also expect to rely on third-party manufacturers or third-party collaborators for the manufacture of commercial supply of BT-11, NX-13 and any other product candidates for which we obtain marketing approval. The facilities used by our CMOs to manufacture our product candidates must be inspected by the FDA or other regulatory authorities after we submit our NDA or comparable marketing application to the FDA or other regulatory authority. We do not have control over a supplier’s or manufacturer’s compliance with laws, regulations and applicable cGMP standards or similar regulatory requirements and other laws and regulations, such as those related to environmental health and safety matters. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or other regulatory authorities, we may be unable to obtain regulatory approval of our marketing applications. In addition, we have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or a comparable foreign regulatory authority finds deficiencies with or does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved.
We may be unable to enter into any agreements with future third-party manufacturers or to do so on acceptable terms. Even if we enter into such agreements, qualifying and validating such manufacturers may take a significant period of time and reliance on third-party manufacturers entails additional risks, including:
|
• |
reliance on the third party for regulatory compliance and quality assurance; |
93
|
• |
the possible breach of the manufacturing agreement by the third party; |
|
• |
the incurrence of upfront scale-up costs prior to commercial approval; |
|
• |
the possible misappropriation of our proprietary information, including our trade secrets and know-how; |
|
• |
the possible increase in costs for the raw materials for our product candidates; and |
|
• |
the possible termination or nonrenewal of any agreement by any third party at a time that is costly or inconvenient for us. |
Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or drugs, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supply of our products.
Our product candidates, and any drugs that we may develop, may compete with other product candidates and drugs for access to manufacturing facilities. The performance of our third-party manufacturers may also be interrupted by production shortages or other supply interruptions resulting from the ongoing COVID-19 pandemic. There are no assurances we would be able to enter into similar commercial arrangements with other manufacturers that operate under cGMP regulations and that might be capable of manufacturing for us in a timely manner. Any performance failure on the part of our existing or future manufacturers could delay clinical development or marketing approval.
We may seek collaborations with third parties for the development or commercialization of our product candidates. If those collaborations are not successful, we may not be able to capitalize on the market potential of these product candidates.
We may seek third-party collaborators for the development and commercialization of our product candidates, including for the commercialization of any of our product candidates that are approved for marketing outside the United States. Our likely collaborators for any such arrangements include regional and national pharmaceutical companies and biotechnology companies. If we enter into any additional such arrangements with any third parties, we will likely have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our product candidates. Our ability to generate revenue from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements.
Collaborations involving our product candidates would pose the following risks to us:
|
• |
collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations; |
|
• |
collaborators may not perform their obligations as expected; |
|
• |
collaborators may not pursue development and commercialization of any product candidates that achieve regulatory approval or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the collaborators’ strategic focus or available funding, or external factors, such as an acquisition, that divert resources or create competing priorities; |
|
• |
collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing; |
|
• |
collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; |
|
• |
product candidates discovered in collaboration with us may be viewed by our collaborators as competitive with their own product candidates or drugs, which may cause collaborators to cease to devote resources to the commercialization of our product candidates; |
|
• |
a collaborator with marketing and distribution rights to one or more of our product candidates that achieve regulatory approval may not commit sufficient resources to the marketing and distribution of such products; |
94
|
• |
disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the preferred course of development, might cause delays or termination of the research, development or commercialization of product candidates, might lead to additional responsibilities for us with respect to product candidates, or might result in litigation or arbitration, any of which would be time-consuming and expensive; |
|
• |
collaborators may not properly maintain or defend our or their intellectual property rights or may use our or their proprietary information in such a way as to invite litigation that could jeopardize or invalidate such intellectual property or proprietary information or expose us to potential litigation; |
|
• |
collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; and |
|
• |
collaborations may be terminated for the convenience of the collaborator and, if terminated, we could be required to raise additional capital to pursue further development or commercialization of the applicable product candidates. |
Collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner or at all. If any future collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program could be delayed, diminished or terminated.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for any collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA or similar regulatory authorities outside the United States, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for our product candidate. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
We may not be able to negotiate additional collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of such product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate revenue.
Risks related to our intellectual property
If we are unable to obtain or protect intellectual property rights related to any of our product candidates, we may not be able to compete effectively in our market.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our product candidates and our LANCE platform. Our success depends in large part on our ability to obtain and maintain patent and other intellectual property protection in the United States and in other countries with respect to our proprietary technology and product candidates.
As of the date of this Annual Report, our patent estate contains at least nine patent families that we own or in-license that protect various aspects of our product candidates. We own or have rights in nine United States patents, 11 United States patent applications, 12 foreign patents, including a European patent that is validated in 33 individual European countries, and at least 47 foreign patent applications. We cannot offer any assurances about which of our patent applications will issue, the breadth of any resulting patent or whether any of the issued patents will be found invalid and unenforceable or will be threatened by third parties. We cannot offer any assurances that
95
the breadth of our granted patents will be sufficient to stop a competitor from developing and commercializing a product, including a biosimilar product that would be competitive with one or more of our product candidates. Furthermore, any successful challenge to these patents or any other patents owned by or licensed to us after patent issuance could deprive us of rights necessary for the successful commercialization of any of our product candidates. Further, if we encounter delays in regulatory approvals, the period of time during which we could market a product candidate under patent protection could be reduced.
The patent prosecution process is expensive and time-consuming. We may not be able to prepare, file and prosecute all necessary or desirable patent applications at a commercially reasonable cost or in a timely manner or in all jurisdictions. It is also possible that we may fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection on them. Moreover, depending on the terms of any future in-licenses to which we may become a party, we may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the patents, covering technology in-licensed from third parties. Therefore, these patents and patent applications may not be prosecuted and enforced in a manner consistent with the best interests of our business.
In addition to the protection provided by our patent estate, we rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not amenable to patent protection. Although we generally require all of our employees to assign their inventions to us, and all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information, or technology to enter into confidentiality agreements, we cannot provide any assurances that all such agreements have been duly executed, or that our trade secrets and other confidential proprietary information will not be disclosed. Moreover, our competitors may independently develop knowledge, methods and know-how equivalent to our trade secrets. Competitors could purchase our products, if approved, and replicate some or all of the competitive advantages we derive from our development efforts for technologies on which we do not have patent protection. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, our agreements or security measures may be breached, and we may not have adequate remedies for any breach. Also, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating the trade secret. In addition, others may independently discover our trade secrets and proprietary information. For example, the FDA is considering whether to make additional information publicly available on a routine basis, including information that we may consider to be trade secrets or other proprietary information, and it is not clear at the present time how the FDA’s disclosure policies may change in the future. If we are unable to prevent material disclosure of the non-patented intellectual property related to our technologies to third parties, and there is no guarantee that we will have any such enforceable trade secret protection, we may not be able to establish or maintain a competitive advantage in our market, which could materially adversely affect our business, results of operations and financial condition.
Patent terms may be inadequate to protect our competitive position on our products for an adequate amount of time, and if we do not obtain protection under the Hatch-Waxman Amendments and similar non-United States legislation for extending the term of patents covering each of our product candidates, our business may be materially harmed.
Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. Depending upon the timing, duration and conditions of FDA marketing approval of our product candidates, one or more of our United States patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Amendments, and similar legislation in the European Union. The Hatch-Waxman Amendments permit a patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval. Only one patent may be extended, and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended.
96
However, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is less than we request, the period during which we can enforce our patent rights for that product will be shortened and our competitors may obtain approval to market competing products sooner. As a result, our revenue from applicable products could be reduced and could have a material adverse effect on our business.
If we fail to comply with our obligations in any future intellectual property licenses with third parties, we could lose rights that are important to our business.
Although we do not currently rely upon any licenses to certain patent rights and proprietary technology for the development our product candidates or the LANCE platform, we may choose to enter into license agreements in the future. These license agreements may impose diligence, development and commercialization timelines and milestone payment, royalty, insurance and other obligations on us. If we fail to comply with our obligations, our licensors may have the right to terminate such licenses, in which event we might not be able to develop, manufacture or market any product that is covered by the intellectual property we in-license from such licensor and may face other penalties. Such an occurrence would materially adversely affect our business prospects.
Licenses to additional third-party technology and materials that may be required for our development programs may not be available in the future or may not be available on commercially reasonable terms, or at all, which could have a material adverse effect on our business and financial condition. We may require the cooperation of our licensors and any upstream licensor for the prosecution, maintenance and enforcement of the licensed and sublicensed intellectual property relating to relevant product candidates, which may not be forthcoming. Therefore, we cannot be certain that the prosecution, maintenance and enforcement of these patent rights will be in a manner consistent with the best interests of our business. If we or our licensor fail to maintain such patents, or if we or our licensor lose rights to those patents or patent applications, the rights we have licensed may be reduced or eliminated and our right to develop and commercialize any of our product candidates that are the subject of such licensed rights could be adversely affected. In addition to the foregoing, the risks associated with patent rights that we license from third parties will also apply to patent rights we may own in the future. Further, if we fail to comply with our development obligations under our license agreements, we may lose our patent rights with respect to such agreement on a territory-by-territory basis, which would affect our patent rights worldwide.
Termination of any future license agreements would reduce or eliminate our rights under these agreements and may result in our having to negotiate new or reinstated agreements with less favorable terms or cause us to lose our rights under these agreements, including our rights to important intellectual property or technology. Any of the foregoing could prevent us from commercializing relevant product candidates, which could have a material adverse effect on our operating results and overall financial condition.
In addition, intellectual property rights that we in-license in the future may be sublicenses under intellectual property owned by third parties, in some cases through multiple tiers. The actions of our licensors may therefore affect our rights to use our sublicensed intellectual property, even if we are in compliance with all of the obligations under our license agreements. Should our licensors or any of the upstream licensors fail to comply with their obligations under the agreements pursuant to which they obtain the rights that are sublicensed to us, or should such agreements be terminated or amended, our ability to develop and commercialize our product candidates may be materially harmed.
Patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our future patents.
Our ability to obtain patents is highly uncertain because, to date, some legal principles remain unresolved, and there has not been a consistent policy regarding the breadth or interpretation of claims allowed in patents in the United States. Furthermore, the specific content of patents and patent applications that are necessary to support and interpret patent claims is highly uncertain due to the complex nature of the relevant legal, scientific, and factual issues. Changes in either patent laws or interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection.
97
For example, on September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act included a number of significant changes to United States patent law. These included provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. The United States Patent and Trademark Office, or USPTO, has developed new and untested regulations and procedures to govern the full implementation of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, became effective in March 2013. The Leahy-Smith Act has also introduced procedures making it easier for third parties to challenge issued patents, as well as to intervene in the prosecution of patent applications. Finally, the Leahy-Smith Act contained new statutory provisions that require the USPTO to issue new regulations for their implementation, and it may take the courts years to interpret the provisions of the new statute. It is too early to tell what, if any, impact the Leahy-Smith Act will have on the operation of our business and the protection and enforcement of our intellectual property. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our future patents. Further, the United States Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on actions by the United States Congress, the federal courts and the USPTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce patents that we have owned or licensed or that we might obtain in the future. An inability to obtain, enforce, and defend patents covering our proprietary technologies would materially and adversely affect our business prospects and financial condition.
Similarly, changes in patent laws and regulations in other countries or jurisdictions, changes in the governmental bodies that enforce them or changes in how the relevant governmental authority enforces patent laws or regulations may weaken our ability to obtain new patents or to enforce patents that we may obtain in the future. Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. For example, if the issuance in a given country of a patent covering an invention is not followed by the issuance in other countries of patents covering the same invention, or if any judicial interpretation of the validity, enforceability or scope of the claims or the written description or enablement, in a patent issued in one country is not similar to the interpretation given to the corresponding patent issued in another country, our ability to protect our intellectual property in those countries may be limited. Changes in either patent laws or in interpretations of patent laws in the United States and other countries may materially diminish the value of our intellectual property or narrow the scope of our patent protection.
We may be involved in lawsuits to protect or enforce our patents, which could be expensive, time consuming and unsuccessful.
Competitors may infringe the patents we have applied for. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. If we initiate legal proceedings against a third party to enforce a patent covering one of our product candidates, the defendant could counterclaim that the patent covering our product or product candidate is invalid and/or unenforceable. In patent litigation in the United States, counterclaims alleging invalidity and/or unenforceability are common, and there are numerous grounds upon which a third party can assert invalidity or unenforceability of a patent.
In an infringement proceeding, a court may decide that the patent claims we are asserting are invalid and/or unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patent claims do not cover the technology in question. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post grant review, inter partes review and equivalent proceedings in foreign jurisdictions (for example, opposition proceedings). Such proceedings could result in revocation of or amendment to our patents in such a way that they no longer cover our product candidates. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we, our patent counsel, and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our product candidates. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing and could have a material adverse impact on our business.
98
Interference proceedings provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to our patent applications. An unfavorable outcome could require us to cease using the related technology or force us to take a license under the patent rights of the prevailing party, if available. Furthermore, our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions, or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common stock.
We may be unsuccessful in licensing or acquiring intellectual property from third parties that may be required to develop and commercialize our product candidates.
A third party may hold intellectual property, including patent rights that are important or necessary to the development and commercialization of our product candidates. It may be necessary for us to use the patented or proprietary technology of third parties to commercialize our product candidates, in which case we would be required to acquire or obtain a license to such intellectual property from these third parties, and we may be unable to do so on commercially reasonable terms or at all. The licensing or acquisition of third-party intellectual property rights is a competitive area, and several more established companies may pursue strategies to license or acquire third-party intellectual property rights that we may consider attractive or necessary. These established companies may have a competitive advantage over us due to their size, capital resources and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment or at all. If we are unable to successfully obtain rights to required third-party intellectual property rights or maintain the existing intellectual property rights we have, we may have to abandon development of the relevant program or product candidate, which could have a material adverse effect on our business.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain.
As our current and future product candidates progress toward commercialization, the possibility of a patent infringement claim against us increases. We cannot provide any assurance that our current and future product candidates do not infringe other parties’ patents or other proprietary rights, and competitors or other parties may assert that we infringe their proprietary rights in any event. We may become party to, or threatened with, adversarial proceedings or litigation regarding intellectual property rights with respect to our current and future product candidates, including interference or derivation proceedings before the USPTO. Even if we believe such claims are without merit, a court of competent jurisdiction could hold that these third-party patents are valid, enforceable and infringed, which could have a negative impact on our ability to commercialize BT-11, NX-13 or any future product candidates. In order to successfully challenge the validity of any such United States patent in federal court, we would need to overcome a presumption of validity. As this burden is high and requires us to present clear and convincing evidence as to the invalidity of any such United States patent claim, there is no assurance that a court of competent jurisdiction would agree with us and invalidate the claims of any such United States patent. Moreover, given the vast number of patents in our field of technology, we cannot be certain that we do not infringe existing patents or that we will not infringe patents that may be granted in the future.
While we may decide to initiate proceedings to challenge the validity of these or other patents in the future, we may be unsuccessful, and courts or patent offices in the United States and abroad could uphold the validity of any such patent. Furthermore, because patent applications can take many years to issue and may be confidential for 18 months or more after filing, and because pending patent claims can be revised before issuance, there may be applications now pending which may later result in issued patents that may be infringed by the manufacture, use or sale of our product candidates. Regardless of when filed, we may fail to identify relevant third-party patents or patent applications, or we may incorrectly conclude that a third-party patent is invalid or not infringed by our product
99
candidates or activities. If a patent holder believes that one of our product candidates infringes its patent, the patent holder may sue us even if we have received patent protection for our technology. Moreover, we may face patent infringement claims from non-practicing entities that have no relevant drug revenue and against whom our own patent portfolio may thus have no deterrent effect. If a patent infringement suit were threatened or brought against us, we could be forced to stop or delay research, development, manufacturing or sales of the drug or product candidate that is the subject of the actual or threatened suit.
If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue commercializing our product candidates. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if a license can be obtained on acceptable terms, the rights may be non-exclusive, which could give our competitors access to the same technology or intellectual property rights licensed to us. If we fail to obtain a required license, we may be unable to effectively market product candidates based on our technology, which could limit our ability to generate revenue or achieve profitability and possibly prevent us from generating revenue sufficient to sustain our operations. Alternatively, we may need to redesign our infringing products, which may be impossible or require substantial time and monetary expenditure. Under certain circumstances, we could be forced, including by court orders, to cease commercializing our product candidates. In addition, in any such proceeding or litigation, we could be found liable for substantial monetary damages, potentially including treble damages and attorneys’ fees, if we are found to have willfully infringed the patent at issue. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could harm our business. Any claims by third parties that we have misappropriated their confidential information or trade secrets could have a similar negative impact on our business.
The cost to us in defending or initiating any litigation or other proceeding relating to patent or other proprietary rights, even if resolved in our favor, could be substantial, and litigation would divert our management’s attention. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could delay our research and development efforts and limit our ability to continue our operations.
We may be subject to claims that our employees, consultants, or independent contractors have wrongfully used or disclosed confidential information of third parties.
We employ individuals who were previously employed at other biotechnology or biopharmaceutical companies. Although we try to ensure that our employees, consultants and advisors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants, or independent contractors have inadvertently or otherwise used or disclosed confidential information of our employees’ former employers or other third parties. We may also be subject to claims that former employers or other third parties have an ownership interest in our future patents. Litigation may be necessary to defend against these claims. There is no guarantee of success in defending these claims, and even if we are successful, litigation could result in substantial cost and be a distraction to our management and other employees.
We may be subject to claims challenging the inventorship or ownership of our future patents and other intellectual property.
We may also be subject to claims that former employees, collaborators, or other third parties have an ownership interest in our patent applications, our future patents, or other intellectual property. We may be subject to ownership disputes in the future arising, for example, from conflicting obligations of consultants or others who are involved in developing our product candidates. Although it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property that we regard as our own, and we cannot be certain that our agreements with such parties will be upheld in the face of a potential challenge, or that they will not be breached, for which we may not have an adequate remedy. The assignment of intellectual property rights may not be self-executing or the assignment agreements may be breached, and litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
100
Reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
If we rely on third parties to manufacture or commercialize BT-11, NX-13 or any future product candidates, or if we collaborate with additional third parties for the development of BT-11, NX-13 or any future product candidates, we must, at times, share trade secrets with them. We may also conduct joint research and development programs that may require us to share trade secrets under the terms of our research and development partnerships or similar agreements. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, consulting agreements or other similar agreements with our advisors, employees, third-party contractors and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, including our trade secrets. Despite the contractual provisions employed when working with third parties, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, a competitor’s discovery of our trade secrets or other unauthorized use or disclosure could have an adverse effect on our business and results of operations.
In addition, these agreements typically restrict the ability of our advisors, employees, third-party contractors and consultants to publish data potentially relating to our trade secrets. Despite our efforts to protect our trade secrets, we may not be able to prevent the unauthorized disclosure or use of our technical know-how or other trade secrets by the parties to these agreements. Moreover, we cannot guarantee that we have entered into such agreements with each party that may have or have had access to our confidential information or proprietary technology and processes. Monitoring unauthorized uses and disclosures is difficult, and we do not know whether the steps we have taken to protect our proprietary technologies will be effective. If any of the collaborators, scientific advisors, employees, contractors and consultants who are parties to these agreements breaches or violates the terms of any of these agreements, we may not have adequate remedies for any such breach or violation, and we could lose our trade secrets as a result. Moreover, if confidential information that is licensed or disclosed to us by our partners, collaborators, or others is inadvertently disclosed or subject to a breach or violation, we may be exposed to liability to the owner of that confidential information. Enforcing a claim that a third party illegally obtained and is using our trade secrets, like patent litigation, is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets.
We may enjoy only limited geographical protection with respect to certain patents and we may not be able to protect our intellectual property rights throughout the world.
Filing and prosecuting patent applications and defending patents covering our product candidates in all countries throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, but enforcement rights are not as strong as that in the United States or Europe. These products may compete with our product candidates, and our future patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
In addition, we may decide to abandon national and regional patent applications before they are granted. The examination of each national or regional patent application is an independent proceeding. As a result, patent applications in the same family may issue as patents in some jurisdictions, such as in the United States, but may issue as patents with claims of different scope or may even be refused in other jurisdictions. It is also quite common that depending on the country, the scope of patent protection may vary for the same product candidate or technology.
While we intend to protect our intellectual property rights in our expected significant markets, we cannot ensure that we will be able to initiate or maintain similar efforts in all jurisdictions in which we may wish to market our product candidates. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate, which may have an adverse effect on our ability to successfully commercialize our product candidates in all of our expected significant foreign markets. If we encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished, and we may face additional competition from others in those jurisdictions.
101
The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws or rules and regulations in the United States and Europe, and many companies have encountered significant difficulties in protecting and defending such rights in such jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets, and other intellectual property rights, which could make it difficult for us to stop the infringement of our future patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in other jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our future patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing as patents, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Some countries also have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, some countries limit the enforceability of patents against government agencies or government contractors. In those countries, the patent owner may have limited remedies, which could materially diminish the value of such patents. If we are forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment, and other requirements imposed by government patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other government fees on patents and/or applications will be due to be paid to the USPTO and various government patent agencies outside of the United States over the lifetime of our patents and/or applications and any patent rights we may obtain in the future. Furthermore, the USPTO and various non-United States government patent agencies require compliance with several procedural, documentary, fee payment and other similar provisions during the patent application process. In many cases, an inadvertent lapse of a patent or patent application can be cured by payment of a late fee or by other means in accordance with the applicable rules. There are situations, however, in which non-compliance can result in abandonment or lapse of the patents or patent applications, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, potential competitors might be able to enter the market, which could have a material adverse effect on our business.
Any trademarks we have obtained or may obtain may be infringed or otherwise violated, or successfully challenged, resulting in harm to our business.
We expect to rely on trademarks as one means to distinguish our product candidates, if approved for marketing, from the drugs of our competitors. Once we select new trademarks and apply to register them, our trademark applications may not be approved. Third parties may oppose or attempt to cancel our trademark applications or trademarks, or otherwise challenge our use of the trademarks. In the event that our trademarks are successfully challenged, we could be forced to rebrand our drugs, which could result in loss of brand recognition and could require us to devote resources to advertising and marketing new brands. Our competitors may infringe or otherwise violate our trademarks and we may not have adequate resources to enforce our trademarks. Any of the foregoing events may have a material adverse effect on our business.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. The following examples are illustrative:
|
• |
others may be able to make products that are similar to or otherwise competitive with our product candidates but that are not covered by the claims of our current or future patents; |
|
• |
an in-license necessary for the manufacture, use, sale, offer for sale or importation of one or more of our product candidates may be terminated by the licensor; |
102
|
• |
we or future collaborators might not have been the first to make the inventions covered by our issued or future issued patents or our pending patent applications; |
|
• |
we or future collaborators might not have been the first to file patent applications covering certain of our inventions; |
|
• |
others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
|
• |
it is possible that our pending patent applications will not lead to issued patents; |
|
• |
issued patents that we own or in-license may be held invalid or unenforceable as a result of legal challenges by our competitors; |
|
• |
issued patents that we own or in-license may not provide coverage for all aspects of our product candidates in all countries; |
|
• |
our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
|
• |
we may not develop additional proprietary technologies that are patentable; and |
|
• |
the patents of others may have an adverse effect on our business. |
Should any of these events occur, they could significantly harm our business, results of operations and prospects.
Risks related to legal and regulatory compliance matters
Our relationships with customers, healthcare providers and third-party payors are subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws, and other healthcare laws and regulations. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
Healthcare providers and third-party payors in the United States and elsewhere will play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our current and future arrangements with healthcare professionals, principal investigators, consultants, customers and third-party payors subject us to various federal and state fraud and abuse laws and other healthcare laws, including, without limitation, the federal Anti-Kickback Statute, the federal civil and criminal false claims laws and transparency laws, including the law commonly referred to as the Physician Payments Sunshine Act, and regulations promulgated under such laws. These laws will impact, among other things, our clinical research, proposed sales, marketing and educational programs, and other interactions with healthcare professionals. The laws that will affect our operations include, but are not limited to:
|
• |
the federal Anti-Kickback Statute, which prohibits, among other things, individuals or entities from knowingly and willfully soliciting, receiving, offering or paying any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind in return for, or to induce, either the referral of an individual, or the purchase, lease, order or arrangement for or recommendation of the purchase, lease, order or arrangement for any good, facility, item or service for which payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. The term “remuneration” has been broadly interpreted to include anything of value. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor. A person does not need to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation; |
|
• |
the federal civil and criminal false claims laws, including, without limitation, the civil False Claims Act, which can be enforced by private citizens through civil whistleblower or qui tam actions, and civil monetary penalty laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment or approval from the federal government, including Medicare, Medicaid and other government payors, that are false or fraudulent or knowingly making, using or causing to be made or used a false |
103
|
record or statement material to a false or fraudulent claim or to avoid, decrease or conceal an obligation to pay money to the federal government. A claim includes “any request or demand” for money or property presented to the United States federal government. Several pharmaceutical and other healthcare companies have been prosecuted under these laws for, among other things, allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of products for unapproved, and thus non-reimbursable, uses. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act; |
|
• |
the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal statutes which prohibit, among other things, a person from knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
|
• |
the federal transparency laws, including the federal Physician Payments Sunshine Act, which requires certain manufacturers of drugs, medical devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services, or CMS, information related to: (1) payments or other “transfers of value’’ made during the previous year to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), and teaching hospitals, and (2) ownership and investment interests held by such physicians and their immediate family members, and, beginning in 2022, will also require applicable manufacturers to report information related to payments and other transfers of value provided in the previous year to physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists, and certified nurse midwives; and |
|
• |
state and foreign law equivalents of each of the above federal laws and regulations; state laws that require manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers, marketing expenditures or drug pricing; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or that otherwise restrict payments that may be made to healthcare providers; and state and local laws that require the registration of pharmaceutical sales representatives. |
Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant penalties, including, without limitation, civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from participating in federal and state funded healthcare programs, such as Medicare and Medicaid, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, contractual damages, diminished profits and future earnings, reputational harm and the curtailment or restructuring of our operations, any of which could harm our business.
The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. The shifting compliance environment and the need to build and maintain robust and expandable systems to comply with multiple jurisdictions with different compliance and/or reporting requirements increases the possibility that a healthcare company may run afoul of one or more of the requirements.
104
Even if we obtain regulatory approval for BT-11, NX-13 or any future product candidates, they will remain subject to ongoing regulatory oversight.
Even if we obtain any regulatory approval for BT-11, NX-13 or any future product candidates, such product candidates, once approved, will be subject to ongoing regulatory requirements applicable to manufacturing, labeling, packaging, storage, advertising, promoting, sampling, record-keeping and submitting of safety and other post-market information, among other things. Any regulatory approvals that we receive for BT-11, NX-13 or any future product candidates may also be subject to a risk evaluation and mitigation strategy, limitations on the approved indicated uses for which the drug may be marketed or to the conditions of approval, or requirements that we conduct potentially costly post-marketing testing, including Phase 4 trials and surveillance to monitor the quality, safety and efficacy of the drug. An unsuccessful post-marketing study or failure to complete such a study could result in the withdrawal of marketing approval. We will further be required to immediately report any serious and unexpected adverse events and certain quality or production problems with our products to regulatory authorities along with other periodic reports.
Any new legislation addressing drug safety issues could result in delays in product development or commercialization, or increased costs to assure compliance. We will also have to comply with requirements concerning advertising and promotion for our products. Promotional communications with respect to prescription drug products are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved label. As such, we will not be allowed to promote our products for indications or uses for which they do not have approval, commonly known as off-label promotion. Physicians, on the other hand, may prescribe products for off-label uses. Although the FDA and other regulatory agencies do not regulate a physician’s choice of drug treatment made in the physician’s independent medical judgment, they do restrict promotional communications from companies or their sales force with respect to off-label uses of products for which marketing approval has not been issued. However, companies may share truthful and not misleading information that is otherwise consistent with a product’s FDA approved labeling. The holder of an approved NDA must submit new or supplemental applications and obtain prior approval for certain changes to the approved product, product labeling, or manufacturing process. A company that is found to have improperly promoted off-label uses of their products may be subject to significant civil, criminal and administrative penalties.
In addition, drug manufacturers are subject to payment of user fees and continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP requirements and adherence to commitments made in the NDA or foreign marketing application. If we, or a regulatory authority, discover previously unknown problems with a drug, such as adverse events of unanticipated severity or frequency, or problems with the facility where the drug is manufactured or if a regulatory authority disagrees with the promotion, marketing or labeling of that drug, a regulatory authority may impose restrictions relative to that drug, the manufacturing facility or us, including requesting a recall or requiring withdrawal of the drug from the market or suspension of manufacturing.
If we fail to comply with applicable regulatory requirements following approval of BT-11, NX-13 or any future product candidates, a regulatory authority may:
|
• |
issue an untitled letter or warning letter asserting that we are in violation of the law; |
|
• |
seek an injunction or impose administrative, civil or criminal penalties or monetary fines; |
|
• |
suspend or withdraw regulatory approval; |
|
• |
suspend any ongoing clinical trials; |
|
• |
refuse to approve a pending NDA or comparable foreign marketing application (or any supplements thereto) submitted by us or our strategic partners; |
|
• |
restrict the marketing or manufacturing of the drug; |
|
• |
seize or detain the drug or otherwise require the withdrawal of the drug from the market; |
|
• |
refuse to permit the import or export of product candidates; or |
|
• |
refuse to allow us to enter into supply contracts, including government contracts. |
105
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize BT-11, NX-13 or any future product candidates and harm our business, financial condition, results of operations and prospects.
Healthcare legislative or regulatory reform measures may have a negative impact on our business and results of operations.
In the United States and some foreign jurisdictions, there have been, and continue to be, several legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of product candidates, restrict or regulate post-approval activities, and affect our ability to profitably sell any product candidates for which we obtain marketing approval.
Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. In March 2010, the ACA was passed, which substantially changed the way healthcare is financed by both the government and private insurers, and significantly impacts the United States pharmaceutical industry. The ACA, among other things: (1) established an annual, nondeductible fee on any entity that manufactures or imports certain specified branded prescription drugs and biologic agents apportioned among these entities according to their market share in some government healthcare programs; (2) expanded the entities eligible for discounts under the 340B drug pricing program; (3) increased the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13% of the average manufacturer price for most branded and generic drugs, respectively, and capped the total rebate amount for innovator drugs at 100% of the Average Manufacturer Price, or AMP; (4) expanded the eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability; (5) addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for certain drugs and biologics that are inhaled, infused, instilled, implanted or injected; (6) introduced a new Medicare Part D coverage gap discount program in which manufacturers must now agree to offer 70% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; (7) created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; and (8) established a Center for Medicare and Medicaid Innovation at CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending.
There remain judicial and Congressional challenges to certain aspects of the ACA, as well as efforts by the Trump administration to repeal or replace certain aspects of the ACA. While Congress has not passed comprehensive repeal legislation, several bills affecting the implementation of certain taxes under the ACA have been signed into law. The Tax Cuts and Jobs Act of 2017, or Tax Act, includes a provision that repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” In addition, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminates the health insurer tax. The Bipartisan Budget Act of 2018, or the BBA, among other things, amended the ACA, effective January 1, 2019, to close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole.” In December 2018, CMS published a new final rule permitting further collections and payments to and from certain ACA qualified health plans and health insurance issuers under the ACA risk adjustment program in response to the outcome of federal district court litigation regarding the method CMS uses to determine this risk adjustment. On April 27, 2020, the United States Supreme Court reversed a Federal Circuit decision that previously upheld Congress’ denial of $12 billion in “risk corridor” funding. On December 14, 2018, a Texas United States District Court Judge ruled that the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress as part of the Tax Act. Additionally, on December 18, 2019, the United States Court of Appeals for the 5th Circuit upheld the District Court ruling that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. On March 2, 2020, the United States Supreme Court granted the petitions for writs of certiorari and held oral argument on November 10, 2020. The case is expected to be decided by mid-2021. It is unclear how such litigation, and other efforts to repeal, replace or challenge the ACA will impact the ACA and our business.
106
Other legislative changes have been proposed and adopted since the ACA was enacted. These changes include aggregate reductions to Medicare payments to providers of 2% per fiscal year pursuant to the Budget Control Act of 2011, which began in 2013, and due to subsequent legislative amendments to the statute, will remain in effect through 2030, with the exception of a temporary suspension from May 1, 2020 through March 31, 2021, unless additional Congressional action is taken. In addition, the American Taxpayer Relief Act of 2012, among other things, further reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These laws may result in additional reductions in Medicare and other healthcare funding, which could have an adverse effect on customers for our product candidates, if approved, and, accordingly, our financial operations.
Additionally, there has been heightened governmental scrutiny in the United States of pharmaceutical pricing practices in light of the rising cost of prescription drugs and biologics. Such scrutiny has resulted in several recent congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products. At the federal level, the Trump administration’s budget proposal for fiscal year 2021 includes a $135 billion allowance to support legislative proposals seeking to reduce drug prices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. On March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare Part D beneficiary out-of-pocket pharmacy expenses, provide an option to cap Medicare Part D beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases. Further, the Trump administration previously released a “Blueprint” to lower drug prices and reduce out of pocket costs of drugs that contained proposals to increase drug manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products, and reduce the out of pocket costs of drug products paid by consumers. On July 24, 2020 and September 13, 2020, President Trump announced a number of executive orders related to prescription drug pricing that collectively attempt to implement several of the administration’s proposals. Further, on November 20, 2020, HHS finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harbor for fixed fees to pharmacy benefit managers for certain services rendered to manufacturers. The probability of success of the other recently announced policies under the Trump administration, which would require additional authorization to be effective, and their impact on our products, if approved, is uncertain, particularly in light of the new incoming Biden administration. The FDA also recently released a final rule, effective November 30, 2020, implementing a portion of President Trump’s Executive Order announced on July 24, 2020 that directed HHS to finalize the Canadian drug importation proposed rule previously issued by HHS and make other changes allowing for personal importation of drugs from Canada. The FDA final rule provides guidance for states to build and submit importation plans for drugs from Canada. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
We expect that these and other healthcare reform measures that may be adopted in the future may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved drug. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our drugs. It is also possible that additional governmental action is taken to address the COVID-19 pandemic.
107
In addition, FDA regulations and guidance may be revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. The Trump administration also took several executive actions, including the issuance of a number of Executive Orders, that could impose significant burdens on, or otherwise materially delay, the FDA’s ability to engage in routine oversight activities such as implementing statutes through rulemaking, issuance of guidance, and review and approval of marketing applications. It is difficult to predict whether or how these requirements will be interpreted and implemented or whether they will be rescinded or replaced under a Biden administration. If executive actions impose restrictions on the FDA’s ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted. Any new regulations or guidance, or revisions or reinterpretations of existing regulations or guidance, may impose additional costs or lengthen FDA review times for BT-11, NX-13 or any future product candidates. We cannot determine how changes in regulations, statutes, policies, or interpretations when and if issued, enacted or adopted, may affect our business in the future. Such changes could, among other things, require:
|
• |
additional clinical trials to be conducted prior to obtaining approval; |
|
• |
changes to manufacturing methods; |
|
• |
recalls, replacements, or discontinuance of one or more of our products; and |
|
• |
additional recordkeeping. |
Such changes would likely require substantial time and impose significant costs, or could reduce the potential commercial value of BT-11, NX-13 or other product candidates, and could materially harm our business and our financial results. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any other products would harm our business, financial condition, and results of operations. Further, we cannot predict the likelihood, nature, or extent of healthcare reform initiatives that may arise from future legislation or administrative action, particularly as a result of the recent U.S. presidential election.
Risks related to employee matters and managing our growth
Our future success depends on our ability to retain key executives and to attract, retain and motivate qualified personnel.
We are highly dependent on the management, development, clinical, financial and business development expertise of our executive officers, particularly Dr. Josep Bassaganya-Riera, our Chairman, President and Chief Executive Officer. Each of our executive officers may currently terminate their employment with us at any time. We do not maintain “key person” insurance for any of our executives or employees.
Recruiting and retaining qualified scientific and clinical personnel and, if we progress the development of our product pipeline toward scaling up for commercialization, manufacturing and sales and marketing personnel, will also be critical to our success. The loss of the services of our executive officers or other key employees could impede the achievement of our development and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize products. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our growth strategy will be limited.
108
We expect to expand our clinical development and regulatory capabilities and potentially implement sales, marketing and distribution capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
As of December 31, 2020, we had 33 employees. As our development progresses, we expect to experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of clinical product development, regulatory affairs and, if any of our product candidates receives marketing approval, sales, marketing and distribution. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Due to our limited financial resources and the limited experience of our management team in managing a company with such anticipated growth, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
Our employees, independent contractors, consultants, commercial collaborators, principal investigators, CROs, suppliers and vendors may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, consultants, commercial collaborators, principal investigators, CROs, suppliers and vendors may engage in fraudulent conduct or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or disclosure of unauthorized activities to us that violates FDA regulations, including those laws requiring the reporting of true, complete and accurate information to the FDA, manufacturing standards, federal and state healthcare laws and regulations, and laws that require the true, complete and accurate reporting of financial information or data. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Misconduct by these parties could also involve the improper use of individually identifiable information, including, without limitation, information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a code of business conduct and ethics, but it is not always possible to identify and deter misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant civil, criminal and administrative penalties, including, without limitation, damages, fines, disgorgement, imprisonment, exclusion from participation in government healthcare programs, such as Medicare and Medicaid, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, and the curtailment or restructuring of our operations.
Risks Related to Ownership of our Common Stock
An active trading market for our common stock may not continue to be developed or sustained.
Prior to our initial public offering, there was no public market for our common stock. Although our common stock is listed on The Nasdaq Global Market, an active trading market for our shares may never develop or be sustained. If an active market for our common stock does not develop or is not sustained, it may be difficult for you to sell shares of our common stock at an attractive price or at all.
The trading price of the shares of our common stock may be volatile, and purchasers of our common stock could incur substantial losses.
Our stock price may be volatile. The stock market in general and the market for biopharmaceutical companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, investors may not be able to sell their common stock at or above the price paid for the shares. The market price for our common stock may be influenced by many factors, including:
|
• |
the commencement, enrollment or results of our clinical trials of BT-11 or NX-13 or any future clinical trials we may conduct, or changes in the development status of our product candidates; |
109
|
• |
any delay in our regulatory filings for BT-11, NX-13 or any other product candidate we may develop, and any adverse development or perceived adverse development with respect to the applicable regulatory authority’s review of such filings, including without limitation the FDA’s issuance of a “refusal to file” letter or a request for additional information; |
|
• |
the success of competitive products or announcements by potential competitors of their product development efforts; |
|
• |
adverse results from, delays in or termination of clinical trials; |
|
• |
adverse regulatory decisions, including failure to receive regulatory approval of our product candidates; |
|
• |
developments or disputes concerning patent applications, issued patents or other proprietary rights; |
|
• |
unanticipated serious safety concerns related to the use of BT-11, NX-13 or any other product candidate; |
|
• |
changes in financial estimates by us or by any equity research analysts who might cover our stock; |
|
• |
conditions or trends in our industry; |
|
• |
changes in the market valuations of similar companies; |
|
• |
stock market price and volume fluctuations of comparable companies and, in particular, those that operate in the biopharmaceutical industry; |
|
• |
publication of research reports about us or our industry or positive or negative recommendations or withdrawal of research coverage by securities analysts; |
|
• |
announcements by us or our competitors of significant acquisitions, strategic collaborations, joint ventures, capital commitments or divestitures; |
|
• |
announcements of investigations or regulatory scrutiny of our operations or lawsuits filed against us; |
|
• |
investors’ general perception of our company and our business; |
|
• |
recruitment or departure of key personnel; |
|
• |
overall performance of the equity markets; |
|
• |
trading volume of our common stock; |
|
• |
sales of common stock by us, our insiders or our other stockholders; |
|
• |
expiration of market stand-off or lock-up agreements; |
|
• |
disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
|
• |
significant lawsuits, including patent or stockholder litigation; |
|
• |
changes in the structure of healthcare payment systems; |
|
• |
general political and economic conditions; and |
|
• |
other events or factors, many of which are beyond our control. |
The stock market in general, and the Nasdaq Global Market and biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies, including very recently in connection with the ongoing COVID-19 pandemic, which has resulted in decreased stock prices for many companies notwithstanding the lack of a fundamental change in their underlying business models or prospects. Broad market and industry factors, including potentially worsening economic conditions and other adverse effects or developments relating to the ongoing COVID-19 pandemic, may negatively affect the market price of our common stock, regardless of our actual operating performance. The realization of any of the above risks or any of a broad range of other risks, including those described in this section, could have a significant and material adverse impact on the market price of our common stock.
110
In addition, in the past, stockholders have initiated class action lawsuits against pharmaceutical and biotechnology companies following periods of volatility in the market prices of these companies’ stock. Such litigation, if instituted against us, could cause us to incur substantial costs and divert management’s attention and resources from our business.
If equity research analysts do not publish research or reports, or publish unfavorable research or reports, about us, our business or our market, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that equity research analysts publish about us and our business. As a newly public company, we have only limited research coverage by equity research analysts. Equity research analysts may elect not to provide research coverage of our common stock, and such lack of research coverage may adversely affect the market price of our common stock. In the event we do have equity research analyst coverage, we will not have any control over the analysts or the content and opinions included in their reports. The price of our stock could decline if one or more equity research analysts downgrade our stock or issue other unfavorable commentary or research. If one or more equity research analysts ceases coverage of our company or fails to publish reports on us regularly, demand for our stock could decrease, which in turn could cause our stock price or trading volume to decline.
A significant portion of our total outstanding shares are restricted from immediate resale but may be sold into the market in the near future. This could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market could occur at any time. If our stockholders sell, or the market perceives that our stockholders intend to sell, substantial amounts of our common stock in the public market, the market price of our common stock could decline significantly.
As of March 29, 2021, we have outstanding 40,117.598 shares of common stock. Of these shares, 6,250,000 shares are freely tradable.
In addition, we intend to file a registration statement on Form S-8 under the Securities Act of 1933, as amended, or the Securities Act, registering the issuance of approximately 5.5 million shares of common stock subject to options or other equity awards issued or reserved for future issuance under our equity incentive plans. Shares registered under this registration statement on Form S-8 will be available for sale in the public market subject to vesting arrangements and exercise of options, the lock-up agreements described above and the restrictions of Rule 144 in the case of our affiliates.
Additionally, the holders of an aggregate of approximately 20.5 million shares of our common stock, or their transferees, will have rights, subject to some conditions, to require us to file one or more registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other stockholders. If we were to register the resale of these shares, they could be freely sold in the public market. If these additional shares are sold, or if it is perceived that they will be sold, in the public market, the trading price of our common stock could decline.
Provisions in our corporate charter documents and under Delaware law may prevent or frustrate attempts by our stockholders to change our management and hinder efforts to acquire a controlling interest in us, and the market price of our common stock may be lower as a result.
There are provisions in our certificate of incorporation and bylaws that may make it difficult for a third party to acquire, or attempt to acquire, control of our company, even if a change of control was considered favorable by you and other stockholders. For example, our board of directors has the authority to issue up to 10,000,000 shares of preferred stock. The board of directors can fix the price, rights, preferences, privileges, and restrictions of the preferred stock without any further vote or action by our stockholders. The issuance of shares of preferred stock may delay or prevent a change of control transaction. As a result, the market price of our common stock and the voting and other rights of our stockholders may be adversely affected. An issuance of shares of preferred stock may result in the loss of voting control to other stockholders.
Our charter documents also contain other provisions that could have an anti-takeover effect, including:
|
• |
only one of our three classes of directors will be elected each year; |
|
• |
stockholders will not be entitled to remove directors other than by a 66 2/3% vote and only for cause; |
|
• |
stockholders will not be permitted to take actions by written consent; |
111
|
• |
stockholders cannot call a special meeting of stockholders; and |
|
• |
stockholders must give advance notice to nominate directors or submit proposals for consideration at stockholder meetings. |
In addition, we are subject to the anti-takeover provisions of Section 203 of the Delaware General Corporation Law, which regulates corporate acquisitions by prohibiting Delaware corporations from engaging in specified business combinations with particular stockholders of those companies. These provisions could discourage potential acquisition proposals and could delay or prevent a change of control transaction. They could also have the effect of discouraging others from making tender offers for our common stock, including transactions that may be in your best interests. These provisions may also prevent changes in our management or limit the price that investors are willing to pay for our stock.
Concentration of ownership of our common stock among our existing executive officers, directors and principal stockholders may prevent new investors from influencing significant corporate decisions.
Our executive officers, directors and current beneficial owners of 5% or more of our common stock and their respective affiliates beneficially own a majority of our common stock. As a result, these persons, acting together, would be able to significantly influence all matters requiring stockholder approval, including the election and removal of directors, any merger, consolidation, sale of all or substantially all of our assets, or other significant corporate transactions.
Some of these persons or entities may have interests different than yours. For example, because many of these stockholders purchased their shares at prices substantially below the current market price of our common stock and have held their shares for a longer period, they may be more interested in selling our company to an acquirer than other investors, or they may want us to pursue strategies that deviate from the interests of other stockholders.
We are an “emerging growth company” and a “smaller reporting company” and, as a result of the reduced disclosure and governance requirements applicable to emerging growth companies and smaller reporting companies, our common stock may be less attractive to investors.
We are an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and we intend to take advantage of some of the exemptions from reporting requirements that are applicable to other public companies that are not emerging growth companies, including:
|
• |
not being required to comply with the auditor attestation requirements in the assessment of our internal control over financial reporting; |
|
• |
not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; |
|
• |
reduced disclosure obligations regarding executive compensation in our periodic reports, proxy statements and registration statements; and |
|
• |
not being required to hold a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We cannot predict if investors will find our common stock less attractive because we will rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company. We will remain an emerging growth company until December 31, 2025 or, if earlier, (i) the last day of the fiscal year in which we have total annual gross revenue of at least $1.07 billion, (ii) the date on which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700 million as of the prior June 30th, or (iii) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
Even after we no longer qualify as an emerging growth company, we may, under certain circumstances, still qualify as a “smaller reporting company,” which would allow us to take advantage of many of the same exemptions from disclosure requirements, including reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements.
112
We have broad discretion in the use our cash and cash equivalents, including the net proceeds from our initial public offering.
We have broad discretion over the use of our cash and cash equivalents, including the net proceeds from our recent initial public offering. You may not agree with our decisions, and our use of the proceeds may not yield any return on your investment. Our failure to apply our cash and cash equivalents effectively could compromise our ability to pursue our growth strategy and we might not be able to yield a significant return, if any, on our investment of these net proceeds. You will not have the opportunity to influence our decisions on how to use our cash and cash equivalents.
Because we do not anticipate paying any cash dividends on our common stock in the foreseeable future, capital appreciation, if any, will be your sole source of gains and you may never receive a return on your investment.
You should not rely on an investment in our common stock to provide dividend income. We have not declared or paid cash dividends on our common stock to date. We currently intend to retain our future earnings, if any, to fund the development and growth of our business. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future. Investors seeking cash dividends should not purchase our common stock.
Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware and the federal district courts of the United States of America will be the exclusive forums for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware is the exclusive forum for the following types of actions or proceedings under Delaware statutory or common law:
|
• |
any derivative action or proceeding brought on our behalf; |
|
• |
any action asserting a breach of fiduciary duty; |
|
• |
any action asserting a claim against us arising under the Delaware General Corporation Law, our amended and restated certificate of incorporation, or our amended and restated bylaws; |
|
• |
any claim or cause of action seeking to interpret, apply, enforce or determine the validity of our restated certificate or our amended and restated bylaws; |
|
• |
any claim or cause of action as to which the Delaware General Corporation Law confers jurisdiction on the Court of Chancery of the state of Delaware; and |
|
• |
any action asserting a claim against us that is governed by the internal-affairs doctrine. |
This provision would not apply to suits brought to enforce a duty or liability created by the Exchange Act. Furthermore, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all such Securities Act actions. Accordingly, both state and federal courts have jurisdiction to entertain such claims. To prevent having to litigate claims in multiple jurisdictions and the threat of inconsistent or contrary rulings by different courts, among other considerations, our amended and restated certificate of incorporation further provides that the federal district courts of the United States of America will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act. While the Delaware courts have determined that such choice of forum provisions are facially valid, a stockholder may nevertheless seek to bring a claim in a venue other than those designated in the exclusive forum provisions. In such instance, we would expect to vigorously assert the validity and enforceability of the exclusive forum provisions of our amended and restated certificate of incorporation. This may require significant additional costs associated with resolving such action in other jurisdictions and there can be no assurance that the provisions will be enforced by a court in those other jurisdictions.
These exclusive forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers, or other employees, which may discourage lawsuits against us and our directors, officers and other employees. If a court were to find either exclusive-forum provision in our amended and restated certificate of incorporation to be inapplicable or unenforceable in an action, we may incur further significant additional costs associated with resolving the dispute in other jurisdictions, all of which could seriously harm our business.
113
General Risks
We maintain a large quantity of sensitive information, including confidential business and personal information in connection with the conduct of our clinical trials and related to our employees, and we are subject to laws and regulations governing the privacy and security of such information.
In the United States, there are numerous federal and state privacy and data security laws and regulations governing the collection, use, disclosure and protection of personal information, including federal and state health information privacy laws, federal and state security breach notification laws, and federal and state consumer protection laws. Each of these constantly evolving laws can be subject to varying interpretations. In addition, we may obtain health information from third parties (including research institutions from which we obtain clinical trial data) that are subject to privacy and security requirements under HIPAA. Depending on the facts and circumstances, we could be subject to criminal penalties if we knowingly obtain, use, or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA.
Even when HIPAA does not apply, according to the Federal Trade Commission, or the FTC, violating consumers’ privacy rights or failing to take appropriate steps to keep consumers’ personal information secure may constitute unfair acts or practices in or affecting commerce in violation of Section 5(a) of the FTC Act. The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Individually identifiable health information is considered sensitive data that merits stronger safeguards.
Foreign data protection laws, including without limitation, the European Union General Data Protection Regulation or the GDPR, which became effective in May 2018, and member state data protection legislation, may also apply to health-related and other personal information obtained outside of the United States. The GDPR governs the collection, use, disclosure, transfer or other processing of personal data of individuals within the European Economic Area, or the EEA. Among other things, the GDPR imposes strict obligations on the ability to process health-related and other personal data of data subjects in the EEA. The GDPR imposes requirements relating to the consent of the individuals to whom the personal data relates, including detailed notices for clinical trial subjects and investigators. The GDPR also includes certain requirements regarding the security of personal data and notification of data processing obligations or security incidents to appropriate data protection authorities or data subjects as well as requirements for establishing a lawful basis on which personal data can be processed. In addition, the GDPR increases the scrutiny of transfers of personal data from clinical trial sites located in the EEA to the United States and other jurisdictions that the European Commission does not recognize as having “adequate” data protection laws. Recent legal developments in Europe have created complexity and compliance uncertainty regarding certain transfers of personal data from the EEA For example, on July 16, 2020, the Court of Justice of the European Union, or the CJEU, invalidated the EU-US Privacy Shield Framework, or the Privacy Shield, under which personal data could be transferred from the EEA. to United States entities who had self-certified under the Privacy Shield scheme. While the CJEU upheld the adequacy of the standard contractual clauses (a standard form of contract approved by the European Commission as an adequate personal data transfer mechanism, and potential alternative to the Privacy Shield), it made clear that reliance on them alone may not necessarily be sufficient in all circumstances. Use of the standard contractual clauses must now be assessed on a case-by-case basis taking into account the legal regime applicable in the destination country, in particular applicable surveillance laws and rights of individuals. The GDPR imposes substantial fines for breaches and violations (up to the greater of €20 million or 4% of our consolidated annual worldwide gross revenue). The GDPR also confers a private right of action on data subjects and consumer associations to lodge complaints with supervisory authorities, seek judicial remedies and obtain compensation for damages resulting from violations of the GDPR.
Further, following Brexit we will have to comply with the GDPR and separately the GDPR as implemented in the United Kingdom, each regime having the ability to fine up to the greater of €20 million / £17 million or 4% of global turnover. The relationship between the United Kingdom and the European Union in relation to certain aspects of data protection law remains unclear, including how data transfers between European Union member states and the United Kingdom will be treated. These changes may lead to additional compliance costs and could increase our overall risk.
114
Compliance with these and any other applicable privacy and data security laws and regulations is a rigorous and time-intensive process, and we may be required to put in place additional mechanisms ensuring compliance with the new data protection rules. If we fail to comply with any such laws or regulations, we may face significant fines and penalties that could adversely affect our business, financial condition and results of operations. Furthermore, the laws are not consistent, and compliance in the event of a widespread data breach is costly. In addition, states are constantly adopting new laws or amending existing laws, requiring attention to frequently changing regulatory requirements. For example, California enacted the California Consumer Privacy Act, or the CCPA, on June 28, 2018, which took effect on January 1, 2020 and has been dubbed the first “GDPR-like” law in the United States. The CCPA gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing and receive detailed information about how their personal information is used by requiring covered companies to provide new disclosures to California consumers (as that term is broadly defined) and provide such consumers new ways to opt-out of certain sales of personal information. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. The CCPA may increase our compliance costs and potential liability. Some observers have noted that the CCPA could mark the beginning of a trend toward more stringent privacy legislation in the United States. Other states are beginning to pass similar laws. For example, an amendment to Nevada’s privacy laws, which went into effect October 1, 2019, requires us to offer to consumers the right to opt-out of the sale of their personal information.
Our business and operations would suffer in the event of computer system failures, cyberattacks or a deficiency in our cybersecurity.
Despite the implementation of security measures, our internal computer systems, and those of third parties on which we rely, are vulnerable to damage from computer viruses, malware, natural disasters, terrorism, war, telecommunication and electrical failures, cyberattacks or cyber-intrusions over the Internet, attachments to emails, persons inside our organization, or persons with access to systems inside our organization. The risk of a security breach or disruption, particularly through cyberattacks or cyber intrusion, including by computer hackers, “phishing” attacks, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our product development programs. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. The techniques used by cyber criminals change frequently, may not be recognized until launched and can originate from a wide variety of sources, including outside groups such as external service providers, organized crime affiliates, terrorist organizations or hostile foreign governments or agencies. We cannot assure you that our security measures will prevent significant breakdowns, data leakages or breaches in our systems or those of our CROs and other contractors and consultants. As a result of the COVID-19 pandemic, we may face increased cybersecurity risks due to our reliance on internet technology and the number of our employees that are working remotely, which may create additional opportunities for cybercriminals to exploit vulnerabilities. For example, companies have experienced an increase in phishing and social engineering attacks from third parties in connection with the COVID-19 global pandemic. To the extent that any disruption or security breach was to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur material legal claims and liability and damage to our reputation, and the further development of our product candidates could be delayed.
Our business activities will be subject to the Foreign Corrupt Practices Act, or FCPA, and similar anti-bribery and anti-corruption laws.
As we expand our business activities outside of the United States, including our clinical trial efforts, we will be subject to the FCPA and similar anti-bribery or anti-corruption laws, regulations or rules of other countries in which we operate. The FCPA generally prohibits offering, promising, giving, or authorizing others to give anything of value, either directly or indirectly, to a non-United States government official in order to influence official action, or otherwise obtain or retain business. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect the transactions of the corporation and to devise and maintain an adequate system of internal accounting controls. Our business is heavily regulated and therefore involves significant interaction with public officials, including officials of non-United States governments. Additionally, in many other countries, the healthcare providers who prescribe pharmaceuticals are employed by their government, and the purchasers of pharmaceuticals are government entities; therefore, our dealings with these prescribers and purchasers will be subject to regulation under the FCPA. Recently the SEC and Department of Justice have increased their FCPA enforcement activities with respect to biotechnology and pharmaceutical companies. There is no certainty that all of
115
our employees, agents, suppliers, manufacturers, contractors, or collaborators, or those of our affiliates, will comply with all applicable laws and regulations, particularly given the high level of complexity of these laws. Violations of these laws and regulations could result in fines, criminal sanctions against us, our officers, or our employees, the closing down of facilities, including those of our suppliers and manufacturers, requirements to obtain export licenses, cessation of business activities in sanctioned countries, implementation of compliance programs, and prohibitions on the conduct of our business. Any such violations could include prohibitions on our ability to offer our products in one or more countries as well as difficulties in manufacturing or continuing to develop our products, and could materially damage our reputation, our brand, our international expansion efforts, our ability to attract and retain employees, and our business, prospects, operating results, and financial condition.
If we fail to maintain proper and effective internal controls, our ability to produce accurate financial statements on a timely basis could be impaired.
We are subject to the reporting requirements of the Securities Exchange Act of 1934, the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, and the rules and regulations of the stock market on which our common stock is listed. The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures and internal control over financial reporting.
Commencing with our fiscal year ending December 31, 2021, we must perform system and process evaluation and testing of our internal control over financial reporting to allow management to report on the effectiveness of our internal control over financial reporting in our Form 10-K filing for that year, as required by Section 404 of the Sarbanes-Oxley Act. This will require that we incur substantial additional professional fees and internal costs to expand our accounting and finance functions and that we expend significant management efforts. Prior to our initial public offering, we have never been required to test our internal control within a specified period, and, as a result, we may experience difficulty in meeting these reporting requirements in a timely manner.
We may identify weaknesses in our system of internal financial and accounting controls and procedures that could result in a material misstatement of our financial statements. Our internal control over financial reporting will not prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud will be detected.
If we are not able to comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner, or if we are unable to maintain proper and effective internal controls, we may not be able to produce timely and accurate financial statements. If that were to happen, the market price of our stock could decline and we could be subject to sanctions or investigations by the stock exchange on which our common stock is listed, the Securities and Exchange Commission or other regulatory authorities.
Our effective tax rate may fluctuate, and we may incur obligations in tax jurisdictions in excess of accrued amounts.
We are subject to taxation in more than one tax jurisdiction. As a result, our effective tax rate is derived from a combination of applicable tax rates in the various places that we operate. In preparing our financial statements, we estimate the amount of tax that will become payable in each of such places. Nevertheless, our effective tax rate may be different than experienced in the past due to numerous factors, including passage of the newly enacted federal income tax law, changes in the mix of our profitability from jurisdiction to jurisdiction, the results of examinations and audits of our tax filings, our inability to secure or sustain acceptable agreements with tax authorities, changes in accounting for income taxes and changes in tax laws. Any of these factors could cause us to experience an effective tax rate significantly different from previous periods or our current expectations and may result in tax obligations in excess of amounts accrued in our financial statements.
Our ability to use our net operating loss carryforwards and certain other tax attributes to offset future taxable income may be subject to certain limitations.
Our net operating loss carryforwards, or NOLs, and certain other tax attributes could expire unused and be unavailable to offset future income tax liabilities because of their limited duration or because of restrictions under U.S. tax law. Our NOLs generated in the taxable year ended on or before December 31, 2017 are only permitted to be carried forward for 20 taxable years under applicable U.S. federal tax law. As of December 31, 2020, we had U.S. federal and state NOLs of $43.9 million and $43.9 million, respectively. The federal NOLs include $2.1 million that may be used to offset up to 100% of future taxable income and will begin to expire in 2037, unless previously
116
utilized. We have a full valuation allowance for deferred tax assets including NOLs. Under the Tax Act, as modified by the CARES Act, federal net operating losses incurred in taxable years ended after December 31, 2017 and in future years may be carried forward indefinitely, but the deductibility of federal net operating losses generated in taxable years beginning after December 31, 2017, and particularly for tax years beginning after December 31, 2020, is limited.
In addition, under Section 382 of the Code, and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a cumulative change, by value, in our ownership by “5-percent stockholders” that exceeds 50 percentage points over a rolling three-year period, the corporation’s ability to use its pre-change NOLs and other pre-change tax attributes to offset its post-change income or taxes may be limited. We have not determined whether we have undergone an ownership change in the past or as a result of our initial public offering. If an ownership change occurs and our ability to use our net operating loss carryforwards is materially limited, it would harm our future operating results by effectively increasing our future tax obligations.
We will incur increased costs and demands upon management as a result of being a public company.
As a public company listed in the United States, we will incur significant additional legal, accounting and other costs. These additional costs could negatively affect our financial results. In addition, changing laws, regulations and standards relating to corporate governance and public disclosure, including regulations implemented by the SEC and The Nasdaq Stock Market, may increase legal and financial compliance costs and make some activities more time-consuming. These laws, regulations and standards are subject to varying interpretations and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If notwithstanding our efforts to comply with new laws, regulations and standards, we fail to comply, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
Failure to comply with these rules might also make it more difficult for us to obtain some types of insurance, including director and officer liability insurance, and we might be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, on committees of our board of directors or as members of senior management.
Item 1B. Unresolved Staff Comments.
None.
Our principal executive office is located at 1800 Kraft Drive, Suite 216, Blacksburg, Virginia, 24060, where we lease approximately 5,500 square feet of office and lab space under a lease that terminates on July 31, 2021. This space houses the internal research and development efforts with a fully functional wet lab equipped with lab benches, hoods, safety equipment and appropriate plumbing and ventilation to conduct hands-on scientific research; and a vivarium, featuring five animal rooms for internal studies. We also occupy approximately 700 square feet of office space in Ashburn, Virginia, which lease expires June 30, 2021. We believe that our facilities are sufficient to meet our current needs and that suitable additional space will be available as and when needed.
We are not subject to any material legal proceedings. From time to time, we may be involved in various claims and legal proceedings relating to claims arising out of our operations. We are not currently a party to any legal proceedings that, in the opinion of our management, are likely to have a material adverse effect on our business. Regardless of outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Item 4. Mine Safety Disclosures.
Not applicable.
117
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information for Common Stock
Our common stock is listed on The Nasdaq Global Market under the symbol “LABP.”
Holders of Record
As of March 29, 2021, we had 19 holders of record of our common stock, which excludes stockholders whose shares were held in nominee or street name by brokers. The actual number of common stockholders is greater than the number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
Dividend Policy
We have never paid cash dividends on any of our capital stock and currently intend to retain our future earnings, if any, to fund the development and growth of our business.
Use of Proceeds from Initial Public Offering
On February 3, 2021, our Registration Statement on Form S-1, as amended (File No. 333-252083), was declared effective in connection with our initial public offering, pursuant to which we sold an aggregate of 6,250,000 shares of our common stock at a price to the public of $16.00 per share. The joint book-running managers of our initial public offering were J.P. Morgan Securities LLC, Jefferies LLC and SVB Leerink LLC, and Raymond James & Associates, Inc. acted as lead manager. There has been no material change in the planned use of proceeds from our initial public offering as described in our prospectus filed pursuant to Rule 424(b)(4) under the Securities Act with the SEC on February 4, 2021.
Issuer Purchases of Equity Securities
None.
Item 6. Selected Financial Data.
We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and are not required to provide the information required under this item.
118
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and related notes.
Overview
We are a clinical-stage biopharmaceutical company focused on the discovery and development of oral therapeutics for patients with autoimmune diseases that are the first to target new mechanisms of action, including the LANCL2, NLRX1 and PLXDC2 immunometabolic pathways. Our core expertise is in the development of therapeutic candidates that target novel pathways at the interface of immunity and metabolism. Based on our understanding of the role that a cell’s metabolic pathways have on modulating inflammatory responses, we aim to inhibit these inflammatory responses by changing the metabolic processes in target cells. We leverage our proprietary AI-based precision medicine platform, which we refer to as our LANCE platform, to identify novel therapeutic targets based on predictions of immunometabolic function and create therapeutic candidates to engage those targets in areas of unmet medical need. Through our LANCE platform, we have identified seven novel immunometabolic targets and product candidates to date across 14 indications, including ulcerative colitis, or UC, Crohn’s disease, or CD, lupus, rheumatoid arthritis, nonalcoholic steatohepatitis, multiple sclerosis, Alzheimer’s disease, asthma, psoriasis, atopic dermatitis, eosinophilic esophagitis, chronic obstructive pulmonary disease, diabetic neuropathy and type 1 diabetes.
We have completed the induction phase of a Phase 2 clinical trial of our lead product candidate, BT-11, for mild to moderate UC in the United States, Russia and Europe to evaluate the efficacy and safety of BT-11 in UC patients. Data from the induction stage of this Phase 2 trial demonstrated that BT-11 was gut-restricted and well tolerated, with no treatment-related significant adverse events and a similar adverse event profile across placebo and BT-11 groups.
We have a limited operating history. Since our inception in 2017, our operations have focused on developing our clinical and preclinical product candidates and our LANCE platform, organizing and staffing our company, business planning, raising capital, establishing our intellectual property portfolio and conducting clinical trials and preclinical studies. We do not have any product candidates approved for sale and have not generated any revenue from product sales. We have funded our operations primarily through the sale of equity securities. Since inception, we have raised an aggregate of $70.0 million of gross proceeds from the sale of shares of our preferred stock and convertible promissory notes.
Since inception, we have incurred significant operating losses. Our net loss was $30.1 million and $13.5 million for the years ended December 31, 2020 and 2019, respectively. As of December 31, 2020, we had an accumulated deficit of $55.7 million. We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate that our expenses will increase significantly in connection with our ongoing activities, as we:
|
• |
conduct our ongoing and planned clinical trials of BT-11 and NX-13, as well as initiate and complete additional clinical trials, as needed; |
|
• |
pursue regulatory approval of BT-11 and NX-13 for the treatment of UC and CD; |
|
• |
leverage our LANCE platform to discover and develop additional product candidates for the treatment of autoimmune diseases; |
|
• |
scale up our clinical and regulatory capabilities; |
|
• |
establish a commercialization infrastructure and scale up external manufacturing and distribution capabilities to commercialize any product candidates for which we may obtain regulatory approval, including BT-11 and NX-13; |
|
• |
adapt our regulatory compliance efforts to incorporate requirements applicable to marketed products; |
|
• |
maintain, expand and protect our intellectual property portfolio; |
|
• |
hire additional clinical, manufacturing and scientific personnel; |
|
• |
add operational, financial and management information systems and personnel, including personnel to support our product development and planned future commercialization efforts; and |
|
• |
incur additional legal, accounting and other expenses in operating as a public company. |
119
On February 3, 2021, we completed our initial public offering (“IPO”) in which we issued and sold 6,250,000 shares of our common stock at a public offering price of $16 per share. The Company received net proceeds of $91.2 million from the IPO, after deducting underwriters’ discounts and commissions.
Components of our results of operations
Research and development expenses
Research and development expenses consist primarily of costs incurred in connection with our research activities, including our discovery efforts, and the development of our product candidates, and include:
|
• |
salaries, benefits, stock-based compensation and other related costs for personnel engaged in research and development functions; |
|
• |
expenses incurred under agreements with third parties, including CROs and other third parties that conduct research, preclinical activities and clinical trials on our behalf, as well as CMOs that manufacture drug material for use in our clinical trials and preclinical studies; |
|
• |
costs of outside consultants, including their fees, and related travel expenses; |
|
• |
the costs of laboratory supplies and acquiring, developing and manufacturing preclinical and clinical trial supply; and |
|
• |
allocated expenses for rent and maintenance of facilities and other operating costs. |
We expense research and development costs as incurred. We track external development costs by product candidate or development program, but we do not allocate personnel costs, or other internal costs to specific development programs or product candidates.
Research and development activities are central to our business model. Product candidates in later stages of clinical development generally have a higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We expect that our research and development expenses will continue to increase substantially for the foreseeable future and will comprise a larger percentage of our total expenses as we complete our ongoing clinical trials, initiate new clinical trials, continue to discover and develop additional product candidates and prepare regulatory filings for any product candidates that successfully complete clinical development.
The successful development of our product candidates is highly uncertain. At this time, we cannot determine with certainty the duration and costs of our existing and future clinical trials of our product candidates or any other product candidate we may develop or if, when, or to what extent we will generate revenue from the commercialization and sale of any product candidate for which we obtain marketing approval. We may never succeed in obtaining marketing approval for any product candidate. The duration, costs and timing of clinical trials and development of our product candidates and any other product candidate we may develop in the future will depend on a variety of factors, including:
|
• |
per patient trial costs; |
|
• |
the number of patients who enroll in each trial; |
|
• |
the number of trials required for approval; |
|
• |
the number of sites included in the trials; |
|
• |
the countries in which the trials are conducted; |
|
• |
the length of time required to enroll eligible patients; |
|
• |
the drop-out or discontinuation rates of patients; |
|
• |
potential additional safety monitoring requested by regulatory agencies; |
|
• |
the duration of patient participation in the trials and follow-up; |
|
• |
the phase of development of the product candidate; and |
|
• |
the efficacy and safety profile of the product candidate. |
120
Our expenditures are subject to additional uncertainties, including the terms and timing of regulatory approvals, and the expense of filing, prosecuting, defending and enforcing any patent claims or other intellectual property rights. We may never succeed in achieving regulatory approval for our product candidates. We may obtain unexpected results from our clinical trials. We may elect to discontinue, delay or modify clinical trials of our product candidates. A change in the outcome of any of these variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA or another regulatory authority were to require us to conduct clinical trials beyond those that we anticipate will be required for the completion of clinical development of a product candidate, or if we experience significant delays in our clinical trials due to patient enrollment or other reasons, we would be required to expend significant additional financial resources and time on the completion of clinical development.
General and administrative expenses
General and administrative expenses consist primarily of salaries and other related costs for personnel in our executive, finance, business development and administrative functions. General and administrative expenses also include legal fees relating to intellectual property and corporate matters, professional fees for accounting, auditing, tax and consulting services; insurance costs, travel expenses and facility-related expenses, which include direct depreciation costs and allocated expenses for rent and maintenance of facilities and other operating costs.
We expect that our general and administrative expenses will increase in the future as we increase our personnel headcount to support our expanded infrastructure, including the development of a commercialization infrastructure for any product candidates for which we may obtain regulatory approval. We also expect to incur increased expenses associated with being a public company, including costs of accounting, audit, legal, regulatory and tax-related services associated with maintaining compliance with stock exchange and SEC requirements, director and officer insurance costs and investor and public relations costs. We anticipate the additional costs for these services will increase our general and administrative expenses by between $1.0 million and $2.0 million on an annual basis.
Interest expense
Interest expense consists of interest due on our convertible promissory notes that were outstanding during the period prior to the conversion of the notes into Series B convertible preferred stock in August 2019.
Income taxes
Since our inception in January 2017, we have generated cumulative federal and state net operating loss for which we have not recorded any net tax benefit due to uncertainty around utilizing these tax attributes within their respective carryforward periods.
As of December 31, 2020, we had federal net operating loss carryforwards, or NOLs, of $43.9 million and state NOLs of $43.9 million that may be available to offset future taxable income. The federal NOLs include $2.1 million available to reduce 100% of future taxable income, which will begin to expire in 2037, if not utilized, and $41.8 million, which can be carried forward indefinitely. The state NOLs will begin to expire in 2037, if not utilized.
Utilization of the net operating losses and credits may be subject to a substantial annual limitation due to ownership change limitations provided by the Internal Revenue Code of 1986, as amended. The annual limitation may result in the expiration of our net operating losses and credits before we can use them. We have recorded a valuation allowance on our net deferred tax assets, including our deferred tax assets related to our net operating loss and research and development tax credit carryforwards.
Other Income, Net
Other income, net consists of interest income received from marketable securities.
121
Results of operations
Comparison of the years ended December 31, 2020 and 2019
The following table summarizes our results of operations for the years ended December 31, 2020 and 2019:
|
|
|
Year ended |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
|
|
|
(in thousands) |
|
||||
|
Operating expenses |
|
|
|
|
|
|
|
|
|
Research and development |
|
$ |
25,338 |
|
|
$ |
11,812 |
|
|
General and administrative |
|
|
5,338 |
|
|
|
1,478 |
|
|
Total operating expenses |
|
|
30,676 |
|
|
|
13,290 |
|
|
Loss from operations |
|
|
(30,676 |
) |
|
|
(13,290 |
) |
|
Other income (expense); |
|
|
|
|
|
|
|
|
|
Interest expense |
|
|
— |
|
|
|
(304 |
) |
|
Gain (loss) from foreign exchange |
|
|
77 |
|
|
|
(33 |
) |
|
Other income, net |
|
|
455 |
|
|
|
160 |
|
|
Other income (expense), net |
|
|
532 |
|
|
|
(177 |
) |
|
Net loss |
|
$ |
(30,144 |
) |
|
$ |
(13,467 |
) |
Research and development expenses
Research and development expenses were $25.3 million for the year ended December 31, 2020 compared to $11.8 million for the year ended December 31, 2019. The increase of $13.5 million was primarily attributable to increased costs associated with ongoing Phase 2 clinical trial activities for BT-11.
The following table summarizes our research and development expenses by product candidate or development program for the years ended December 31, 2020 and 2019:
|
|
|
Year ended |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
|
|
(in thousands) |
|
|||||
|
BT-11 |
|
$ |
20,752 |
|
|
$ |
10,621 |
|
|
BT-104 |
|
|
104 |
|
|
|
— |
|
|
NX-13 |
|
|
3,746 |
|
|
|
657 |
|
|
PX-69 |
|
|
125 |
|
|
|
— |
|
|
Other discovery pipeline, LANCE platform and unallocated costs |
|
|
611 |
|
|
|
534 |
|
|
Total research and development expenses |
|
$ |
25,338 |
|
|
$ |
11,812 |
|
General and administrative expenses
General and administrative expenses were $5.3 million for the year ended December 31, 2020 compared to $1.5 million for the year ended December 31, 2019. The increase of $3.8 million was primarily attributable to increases in patent costs, related legal fees and other outside professional services.
Other Income, net
Other income, net was $0.5 million for the year ended December 31, 2020 compared to other expense, net of $0.2 million for the year ended December 31, 2019. The increase was due to interest received from marketable securities.
122
Liquidity and capital resources
Since our inception, we have incurred significant operating losses and negative cash flows from our operations. We expect to incur significant expenses and operating losses for the foreseeable future as we advance the preclinical and clinical development of our research programs and product candidates. We expect that our research and development and general and administrative costs will increase in connection with conducting additional preclinical studies and clinical trials for our current and future research programs and product candidates, including BT-11 and NX-13, discovering and developing new product candidates using the LANCE precision medicine platform, contracting with CMOs to support preclinical studies and clinical trials, expanding our intellectual property portfolio, and providing general and administrative support for our operations. As a result, we will need additional capital to fund our operations, which we may obtain from additional equity or debt financings, collaborations, licensing arrangements, or other sources.
We do not currently have any approved products and have never generated any revenue from product sales. To date, we have financed our operations primarily through equity financings. As of December 31, 2020, we had $28.1 million in cash, cash equivalents and marketable securities and an accumulated deficit of $55.7 million. We had no indebtedness as of December 31, 2020. As of December 31, 2019, we had $50.0 million in cash, cash equivalents and marketable securities and an accumulated deficit of $25.6 million. We had no indebtedness as of December 31, 2020 and 2019.
On February 3, 2021, we completed our initial public offering (“IPO”) in which we issued and sold 6,250,000 shares of our common stock at a public offering price of $16 per share. The Company received net proceeds of $91.2 million from the IPO, after deducting underwriters’ discounts and commissions.
The following table summarizes our sources and uses of cash for each of the periods set forth below:
|
|
|
Year ended |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
Net cash used in operating activities |
|
$ |
(22,962 |
) |
|
$ |
(9,874 |
) |
|
Net cash provided by (used in) investing activities |
|
|
14,131 |
|
|
|
(40,325 |
) |
|
Net cash provided by financing activities |
|
|
1,518 |
|
|
|
59,648 |
|
|
Net increase (decrease) in cash and cash equivalents |
|
$ |
(7,313 |
) |
|
$ |
9,449 |
|
Operating activities
Net cash used in operating activities for the year ended December 31, 2020 was $23 million, consisting primarily of our net loss of $30.1 million as we incurred expenses associated with research activities for our lead product candidates and incurred general and administrative expenses. Net cash used in operating activities for the year ended December 31, 2019 was $9.9 million, consisting primarily of our net loss of $13.5 million as we incurred expenses associated with research activities for our lead product candidates and incurred general and administrative expenses.
Investing activities
Net cash provided by investing activities for the year ended December 31, 2020 was $14.1 million, consisting of proceeds from sales and maturities of marketable securities partially offset by purchases of available-for-sale marketable securities. Net cash used in investing activities for the year ended December 31, 2019 was $40.3 million, consisting primarily of purchases of available-for-sale marketable securities.
Financing activities
Net cash provided by financing activities in the year ended December 31, 2020 of $1.5 million was related to proceeds from exercise of stock options. Net cash provided by financing activities in the year ended December 31, 2019 of $59.6 million was related to net proceeds of $51.7 million from the issuance of our Series B convertible preferred stock and $8.0 million from the issuance of our convertible promissory notes that were subsequently converted into Series B convertible preferred stock.
123
Funding requirements
To date, we have not generated any revenues from the commercial sale of approved drug products, and we do not expect to generate substantial revenue for at least the next few years. If we fail to complete the development of our product candidates in a timely manner or fail to obtain their regulatory approval, our ability to generate future revenue will be compromised. We do not know when, or if, we will generate any revenue from our product candidates, and we do not expect to generate significant revenue unless and until we obtain regulatory approval of, and commercialize, our product candidates. We expect our expenses to increase in connection with our ongoing activities, particularly as we continue the research and development of, continue or initiate clinical trials of and seek marketing approval for our product candidates. In addition, if we obtain approval for any of our product candidates, we expect to incur significant commercialization expenses related to sales, marketing, manufacturing and distribution. Furthermore, we expect to incur additional costs associated with operating as a public company. We anticipate that we will need substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research and development programs or future commercialization efforts.
Identifying potential product candidates and conducting preclinical studies and clinical trials is a time-consuming, expensive and uncertain process that takes many years to complete, and we may never generate the necessary data or results required to obtain marketing approval and achieve product sales. In addition, our product candidates, if approved, may not achieve commercial success. Our commercial revenues, if any, will be derived from sales of product candidates that we do not expect to be commercially available in the near term, if at all.
We believe that the existing cash and cash equivalents, will enable us to fund our operating expenses and capital expenditure requirements into 2023. We have based these estimates on assumptions that may prove to be imprecise, and we could utilize our available capital resources sooner than we expect.
Because of the numerous risks and uncertainties associated with research, development and commercialization of pharmaceutical drugs, it is difficult to estimate with certainty the amount of our working capital requirements. Our future funding requirements will depend on many factors, including:
|
• |
the scope, progress, costs and results of our ongoing and planned clinical trials of BT-11 and NX-13; |
|
• |
the costs and results of discovery work using our LANCE precision medicine platform; |
|
• |
the scope, progress, costs and results of preclinical development, laboratory testing and clinical trials for any future product candidates we may decide to pursue; |
|
• |
the extent to which we in-license or acquire rights to other products, product candidates or technologies; |
|
• |
the costs and timing of process development and manufacturing scale-up activities associated with our product candidates and other programs we advance them through preclinical and clinical development; |
|
• |
the number and development requirements of other product candidates that we may pursue; |
|
• |
the costs, timing and outcome of regulatory review of our product candidates; |
|
• |
the costs and timing of future commercialization activities, including product manufacturing, marketing, sales and distribution, for any of our product candidates for which we receive marketing approval; |
|
• |
the revenue, if any, received from commercial sales of our product candidates for which we receive marketing approval; |
|
• |
our ability to establish and maintain strategic collaborations, licensing or other agreements and the financial terms of such agreements; and |
|
• |
the costs and timing of preparing, filing and prosecuting patent applications, maintaining and protecting our intellectual property rights and defending against any intellectual property-related claims. |
Further, our operating results may change in the future, and we may need additional funds to meet operational needs and capital requirements associated with such operating plans.
124
Our future commercial revenue, if any, will be derived from sales of products that we do not expect to be commercially available for several years, if at all. Until such time, if ever, that we can generate product revenue sufficient to achieve profitability, we expect to finance our cash needs through a combination of equity offerings, debt financings, collaboration agreements, other third-party funding, strategic alliances, licensing arrangements and marketing and distribution arrangements. Adequate additional financing may not be available to us on acceptable terms, or at all. We currently have no credit facility or committed sources of capital. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through other third-party funding, collaboration agreements, strategic alliances, licensing arrangements or marketing and distribution arrangements, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market products or product candidates that we would otherwise prefer to develop and market ourselves.
Contractual obligations, commitments and contingencies
The following table summarizes our contractual obligations as of December 31, 2020 and the effects that such obligations are expected to have on our liquidity and cash flows in future periods:
|
|
|
Payments due by period |
|
|||||||||||||||||
|
(in thousands) |
|
Total |
|
|
Less |
|
|
1 to 3 |
|
|
4 to 5 |
|
|
More |
|
|||||
|
Operating lease commitments(1) |
|
$ |
108 |
|
|
$ |
108 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Total |
|
$ |
108 |
|
|
$ |
108 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
(1) |
Amounts in the table reflect payments due for our headquarters in Blacksburg, Virginia under an operating lease agreement that expires in July 2021. |
We enter into contracts in the normal course of business with third-party contract organizations for clinical trials, preclinical studies, manufacturing and other services and products for operating purposes. These contracts generally provide for termination following a certain period after notice and therefore we believe that our non-cancelable obligations under these agreements are not material.
Off-balance sheet arrangements
We have not entered into any off-balance sheet arrangements.
Critical accounting policies and significant judgments and estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with generally accepted accounting principles in the United States. The preparation of our consolidated financial statements and related disclosures requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities, costs and expenses and the disclosure of contingent assets and liabilities in our consolidated financial statements. We base our estimates on historical experience, known trends and events and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. We evaluate our estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are described in more detail in the notes to our consolidated financial statements appearing at the end of this prospectus, we believe that the following accounting policies are those most critical to the judgments and estimates used in the preparation of our consolidated financial statements.
125
Research and development expenses
The majority of our operating expenses to date have been incurred in research and development activities. As part of the process of preparing our consolidated financial statements, we estimate our accrued research and development expenses at each consolidated balance sheet date. This process involves reviewing purchase orders and open contracts, communicating with our personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated costs incurred for the services when we have not yet been invoiced or otherwise notified of the actual cost. The majority of our service providers invoice us monthly in arrears for services performed or when contractual milestones are met; however, some require advance payments. We make estimates of our accrued expenses as of each consolidated balance sheet date in our consolidated financial statements based on facts and circumstances known to us at that time. We periodically confirm the accuracy of our estimates with the service providers and make adjustments as necessary. The significant estimates in our accrued research and development expenses include the costs incurred for services performed by CROs with research and development activities for which we have not yet been invoiced.
We base our expenses related to research and development activities on our estimates of the services received and efforts expended pursuant to quotes and contracts with vendors that conduct research and development on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. There may be instances in which payments made to our vendors will exceed the level of services provided and result in a prepayment of the research and development expense. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we adjust the accrual or prepaid accordingly.
Stock-Based Compensation
We account for share-based compensation awards in accordance with FASB ASC Topic 18, Compensation—Stock Compensation (ASC 718). ASC 718 requires all share-based payments, including grants of stock options, to be recognized in the consolidated statements of operations and comprehensive income (loss) based on their respective fair values.
The fair value of our stock options has been determined using the Black-Scholes option-pricing model. The Black-Sholes option-pricing model requires the input of subjective assumptions, including (i) the expected stock price volatility, (ii) the expected term of the award, (iii) the risk-free interest rate and (iv) expected dividends. Due to the lack of historical and implied volatility data of our common stock, the expected stock price volatility has been estimated based on the historical volatilities of a specified group of companies in our industry for a period equal to the expected life of the option. We selected companies with comparable characteristics, including enterprise value, risk profiles and position within the industry and with historical share price information sufficient to meet the expected term of the stock options. The historical volatility data has been computed using the daily closing prices for the selected companies.
The expected life of the options granted represents the period of time that options granted are expected to be outstanding and is calculated using the simplified method, which is the mid-point between the vesting date and the end of the contractual term for each option. The risk-free interest rate is based on a zero coupon, United States Treasury instrument whose term is consistent with the expected life of the stock option. We have not paid, and do not anticipate paying, cash dividends on our shares of common stock; therefore, the expected dividend yield is zero.
We recognize the grant-date fair value of an award as compensation expense on a straight-line basis over the requisite service period, which typically corresponds to the vesting period for the award. In certain circumstances the amount of compensation cost recognized is adjusted to be at least equal to the portion of the grant-date value of the award that was vested at the balance sheet date. We have elected to account for forfeitures as they occur and, upon forfeiture of an award prior to vesting, we reverse any previously recognized compensation expense related to that award.
126
Quantitative and qualitative disclosures about market risk
Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates, particularly because our cash is held in interest-bearing money market accounts. Due to the short- term maturities of our cash equivalents and the low risk profile of our investments, an immediate 100 basis point change in interest rates would not have a material effect on the fair market value of our cash equivalents.
Inflation generally affects us by increasing our cost of labor and clinical trial costs. We do not believe that inflation had a material effect on our business, financial condition or results of operations during the years ended December 31, 2020 and 2019.
Recent accounting pronouncements
A description of recent accounting pronouncements that may potentially impact our financial position, results of operations or cash flows is disclosed in Note 2 to our audited consolidated financial statements included elsewhere in this prospectus.
Emerging growth company status
We qualify as an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, as amended, or JOBS Act. As an “emerging growth company” we may take advantage of reduced reporting requirements that are otherwise applicable to public companies. These provisions include, but are not limited to:
|
• |
the option to present only two years of audited consolidated financial statements and only two years of related “Management’s discussion and analysis of financial condition and results of operations” in this prospectus; |
|
• |
not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as amended; |
|
• |
not being required to comply with any requirements that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the consolidated financial statements (i.e., an auditor discussion and analysis); |
|
• |
reduced disclosure obligations regarding executive compensation in our periodic reports, proxy statements and registration statements; and |
|
• |
exemptions from the requirements of holding a non-binding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We may take advantage of these provisions until the last day of our fiscal year following the fifth anniversary of the completion of this offering. However, if any of the following events occur prior to the end of such five-year period, (i) our annual gross revenue exceeds $1.07 billion, (ii) we issue more than $1.0 billion of non-convertible debt in any three-year period or (iii) we become a “large accelerated filer,” (as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, or the Exchange Act), we will cease to be an emerging growth company prior to the end of such five-year period. We will be deemed to be a “large accelerated filer” at such time that we (a) have an aggregate worldwide market value of common equity securities held by non-affiliates of $700.0 million or more as of the last business day of our most recently completed second fiscal quarter, (b) have been required to file annual and quarterly reports under the Exchange Act, for a period of at least 12 months and (c) have filed at least one annual report pursuant to the Exchange Act. Even after we no longer qualify as an emerging growth company, we may still qualify as a “smaller reporting company,” which would allow us to take advantage of many of the same exemptions from disclosure requirements including reduced disclosure obligations regarding executive compensation in this prospectus and our periodic reports and proxy statements.
We have elected to take advantage of certain of the reduced disclosure obligations in the registration statement of which this prospectus forms a part and may elect to take advantage of other reduced reporting requirements in future filings. As a result, the information that we provide to our stockholders may be different than you might receive from other public reporting companies in which you hold equity interests.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. We have elected to take advantage of this extended transition period.
127
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and are not required to provide the information required under this item.
Item 8. Financial Statements and Supplementary Data.
128
Landos Biopharma, Inc. and Subsidiaries
Index to consolidated financial statements
|
|
Page |
|
F-2 |
|
|
|
|
|
Consolidated financial statements: |
|
|
F-3 |
|
|
Consolidated statements of operations and comprehensive loss |
F-4 |
|
Consolidated statements of convertible preferred stock and stockholders’ deficit |
F-5 |
|
F-6 |
|
|
F-7 |
F-1
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors of Landos Biopharma, Inc. and Subsidiaries
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Landos Biopharma, Inc. and Subsidiaries (the Company) as of December 31, 2020 and 2019, the related consolidated statements of operations and comprehensive loss, convertible preferred stock and stockholders’ deficit and cash flows for each of the two years in the period ended December 31, 2020, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2020 and 2019, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2020, in conformity with U.S. generally accepted accounting principles.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2020.
Raleigh, North Carolina
March 31, 2021
F-2
Landos Biopharma, Inc. and Subsidiaries
(In thousands, except share and per share amounts)
|
|
|
December 31, |
|
|||||
|
|
|
|
2020 |
|
|
|
2019 |
|
|
Assets |
|
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
2,416 |
|
|
$ |
9,808 |
|
|
Marketable securities, available for sale |
|
|
25,718 |
|
|
|
40,157 |
|
|
Incentive and tax receivables |
|
|
154 |
|
|
|
1 |
|
|
Prepaid expenses and other current assets |
|
|
202 |
|
|
|
144 |
|
|
Deferred offering costs |
|
|
1,398 |
|
|
|
— |
|
|
Total current assets |
|
|
29,888 |
|
|
|
50,110 |
|
|
Property and equipment, net |
|
|
444 |
|
|
|
359 |
|
|
Other assets |
|
|
— |
|
|
|
11 |
|
|
Total assets |
|
$ |
30,332 |
|
|
$ |
50,480 |
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities, convertible preferred stock and stockholders’ (deficit) equity |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
8,606 |
|
|
$ |
2,048 |
|
|
Accrued liabilities |
|
|
1,939 |
|
|
|
978 |
|
|
Other current liabilities |
|
|
489 |
|
|
|
— |
|
|
Total current liabilities |
|
|
11,034 |
|
|
|
3,026 |
|
|
Other liabilities |
|
|
276 |
|
|
|
— |
|
|
Total liabilities |
|
|
11,310 |
|
|
|
3,026 |
|
|
|
|
|
|
|
|
|
|
|
|
Commitments and contingencies (Note 8) |
|
|
|
|
|
|
|
|
|
Convertible preferred stock, $0.01 par value, 11,260,608 shares authorized, issued and outstanding as of December 31, 2020 and 2019; aggregate liquidation preference of $70,254 as of December 31, 2020 and 2019 |
|
|
73,037 |
|
|
|
73,037 |
|
|
|
|
|
|
|
|
|
|
|
|
Stockholders’ (deficit) equity: |
|
|
|
|
|
|
|
|
|
Common stock, $0.01 par value; 37,410,450 shares authorized as of December 31, 2020 and 2019; 12,767,909 and 11,784,147 shares issued and outstanding as of December 31, 2020 and 2019, respectively |
|
|
71 |
|
|
|
63 |
|
|
Additional paid-in capital |
|
|
1,633 |
|
|
|
16 |
|
|
Accumulated other comprehensive gain (loss) |
|
|
10 |
|
|
|
(77 |
) |
|
Accumulated deficit |
|
|
(55,729 |
) |
|
|
(25,585 |
) |
|
Total stockholders’ (deficit) equity |
|
$ |
(54,015 |
) |
|
$ |
(25,583 |
) |
|
Total liabilities, convertible preferred stock and stockholders’ (deficit) equity |
|
$ |
30,332 |
|
|
$ |
50,480 |
|
The accompanying notes are an integral part of these consolidated financial statements.
F-3
Landos Biopharma, Inc. and Subsidiaries
Consolidated statements of operations and comprehensive loss
(In thousands, except share and per share amounts)
|
|
|
Year ended December 31, |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
Research and development |
|
$ |
25,338 |
|
|
$ |
11,812 |
|
|
General and administrative |
|
|
5,338 |
|
|
|
1,478 |
|
|
Total operating expenses |
|
|
30,676 |
|
|
|
13,290 |
|
|
Loss from operations |
|
|
(30,676 |
) |
|
|
(13,290 |
) |
|
Other income (expenses): |
|
|
|
|
|
|
|
|
|
Interest expense |
|
— |
|
|
|
(304 |
) |
|
|
Gain/(loss) from foreign exchange |
|
|
77 |
|
|
|
(33 |
) |
|
Other income, net |
|
|
455 |
|
|
|
160 |
|
|
Other income (expense), net |
|
|
532 |
|
|
|
(177 |
) |
|
Net loss |
|
$ |
(30,144 |
) |
|
$ |
(13,467 |
) |
|
Net loss per share, basic and diluted |
|
$ |
(2.47 |
) |
|
$ |
(1.18 |
) |
|
Weighted-average shares used to compute net loss per share, basic and diluted |
|
|
12,227,823 |
|
|
|
11,393,549 |
|
|
Net loss |
|
$ |
(30,144 |
) |
|
$ |
(13,467 |
) |
|
Unrealized gain/(loss) on available-for-sale securities |
|
|
87 |
|
|
|
(77 |
) |
|
Comprehensive loss |
|
$ |
(30,057 |
) |
|
$ |
(13,544 |
) |
The accompanying notes are an integral part of these consolidated financial statements.
F-4
Landos Biopharma, Inc. and Subsidiaries
Consolidated statements of convertible preferred stock and stockholders’ deficit
(In thousands, except share amounts)
|
|
|
Convertible preferred stock |
|
|
Convertible preferred stock |
|
|
Common stock |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
Shares |
|
|
Amounts |
|
|
Shares |
|
|
Amounts |
|
|
Shares |
|
|
Amounts |
|
|
Additional paid-in capital |
|
|
Tranche right |
|
|
Accumulated deficit |
|
|
Accumulated other comprehensive loss |
|
|
Total stockholders’ deficit |
|
|||||||||||
|
Balance at December 31, 2018 |
|
|
— |
|
|
$ |
— |
|
|
|
3,224,034 |
|
|
$ |
32 |
|
|
|
11,011,417 |
|
|
$ |
59 |
|
|
$ |
10,044 |
|
|
|
— |
|
|
$ |
(9,135 |
) |
|
$ |
— |
|
|
$ |
1,000 |
|
|
Compensation expense related to vesting of common stock issued to Xontogeny (See Note 6) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
772,730 |
|
|
|
4 |
|
|
|
47 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
51 |
|
|
Beneficial conversion option on convertible promissory notes (See Note 5) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
20 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
20 |
|
|
Issuance of Series B convertible preferred stock, net of issuance costs of $267 |
|
|
6,935,086 |
|
|
|
51,668 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Conversion of convertible promissory notes into convertible preferred stock (See Note 5) |
|
|
1,101,488 |
|
|
|
8,279 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(20 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(20 |
) |
|
Reclassification of Series A convertible preferred stock from permanent to mezzanine equity |
|
|
3,224,034 |
|
|
|
13,090 |
|
|
|
(3,224,034 |
) |
|
|
(32 |
) |
|
|
— |
|
|
|
— |
|
|
|
(10,075 |
) |
|
|
— |
|
|
|
(2,983 |
) |
|
|
— |
|
|
|
(13,090 |
) |
|
Unrealized loss on marketable securities |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
(77 |
) |
|
|
(77 |
) |
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(13,467 |
) |
|
|
— |
|
|
|
(13,467 |
) |
|
Balance at December 31, 2019 |
|
|
11,260,608 |
|
|
|
73,037 |
|
|
|
— |
|
|
|
— |
|
|
|
11,784,147 |
|
|
$ |
63 |
|
|
$ |
16 |
|
|
|
— |
|
|
$ |
(25,585 |
) |
|
$ |
(77 |
) |
|
$ |
(25,583 |
) |
|
Compensation expense related to vesting of common stock issued to Xontogeny (See Note 6) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
579,548 |
|
|
|
4 |
|
|
|
35 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
39 |
|
|
Stock option exercise |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
404,214 |
|
|
|
4 |
|
|
|
749 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
753 |
|
|
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
833 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
833 |
|
|
Unrealized gain on marketable securities |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
87 |
|
|
|
87 |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(30,144 |
) |
|
|
— |
|
|
|
(30,144 |
) |
|
Balance at December 31, 2020 |
|
|
11,260,608 |
|
|
$ |
73,037 |
|
|
|
|
|
|
$ |
|
|
|
|
12,767,909 |
|
|
$ |
71 |
|
|
$ |
1,633 |
|
|
$ |
|
|
|
$ |
(55,729 |
) |
|
$ |
10 |
|
|
$ |
(54,015 |
) |
The accompanying notes are an integral part of these consolidated financial statements.
F-5
Landos Biopharma, Inc. and Subsidiaries
Consolidated statements of cash flows
(In thousands)
|
|
|
Year ended December 31, |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
CASH FLOWS FROM OPERATING ACTIVITIES: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(30,144 |
) |
|
$ |
(13,467 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
Compensation expense related to vesting of common stock issued to Xontogeny |
|
39 |
|
|
51 |
|
||
|
Depreciation of property and equipment |
|
137 |
|
|
103 |
|
||
|
Accrued interest on convertible promissory notes |
|
|
— |
|
|
259 |
|
|
|
Accrued interest on marketable securities |
|
9 |
|
|
|
(67 |
) |
|
|
Stock-based compensation expense |
|
833 |
|
|
|
— |
|
|
|
Net realized gain/(loss) on sale of marketable securities |
|
1 |
|
|
|
— |
|
|
|
Net (accretion of discount) amortization of premium on marketable securities |
|
213 |
|
|
|
— |
|
|
|
Unrealized gain/(loss) from foreign exchange |
|
79 |
|
|
|
(70 |
) |
|
|
Other |
|
|
— |
|
|
40 |
|
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
Incentive and tax receivables |
|
|
(153 |
) |
|
|
2,091 |
|
|
Prepaid expenses and other assets |
|
|
(643 |
) |
|
|
(103 |
) |
|
Accounts payable |
|
|
6,506 |
|
|
415 |
|
|
|
Other liabilities |
|
161 |
|
|
874 |
|
||
|
Net cash (used in) operating activities |
|
|
(22,962 |
) |
|
|
(9,874 |
) |
|
CASH FLOWS FROM INVESTING ACTIVITIES: |
|
|
|
|
|
|
|
|
|
Purchase of property and equipment |
|
|
(181 |
) |
|
|
(158 |
) |
|
Purchase of available-for-sale marketable securities |
|
|
(21,718 |
) |
|
|
(40,319 |
) |
|
Proceeds from sales and maturities of marketable securities |
|
|
36,030 |
|
|
152 |
|
|
|
Net cash provided by (used in) investing activities |
|
|
14,131 |
|
|
|
(40,325 |
) |
|
CASH FLOWS FROM FINANCING ACTIVITIES: |
|
|
|
|
|
|
|
|
|
Proceeds from issuance of Series B convertible preferred stock, net of issuance costs of $267 |
|
|
— |
|
|
|
51,668 |
|
|
Proceeds from issuance of convertible promissory notes, net of issuance costs of $20 |
|
|
— |
|
|
|
7,980 |
|
|
Proceeds from exercise of stock options |
|
|
1,518 |
|
|
|
— |
|
|
Net cash provided by financing activities |
|
|
1,518 |
|
|
|
59,648 |
|
|
Net change in cash and cash equivalents |
|
|
(7,313 |
) |
|
|
9,449 |
|
|
Cash and cash equivalents at beginning of period |
|
|
9,808 |
|
|
359 |
|
|
|
Effect of exchange rates on cash |
|
|
(79 |
) |
|
|
— |
|
|
Cash and cash equivalents at end of period |
|
$ |
2,416 |
|
|
$ |
9,808 |
|
|
SUPPLEMENTAL DISCLOSURE OF NONCASH FINANCING ITEM: |
|
|
|
|
|
|
|
|
|
Cash paid for interest |
|
$ |
— |
|
|
$ |
27 |
|
|
NONCASH INVESTING AND FINANCING ACTIVITY: |
|
|
|
|
|
|
|
|
|
Conversion of convertible promissory notes and related interest to Series B preferred stock |
|
$ |
— |
|
|
$ |
8,259 |
|
|
Purchase of fixed assets in accounts payable |
|
$ |
41 |
|
|
$ |
— |
|
|
Deferred offering costs included in accounts payable and accrued liabilities |
|
$ |
811 |
|
|
$ |
— |
|
|
Unrealized loss on available-for-sale marketable securities |
|
$ |
87 |
|
|
$ |
77 |
|
|
Beneficial conversion feature discount on convertible promissory notes |
|
$ |
— |
|
|
$ |
20 |
|
The accompanying notes are an integral part of these consolidated financial statements.
F-6
Landos Biopharma, Inc. and Subsidiaries
Notes to consolidated financial statements
As of and for the years ended December 31, 2020, 2019 and 2018
1. Organization and description of the business
Description of business
Landos Biopharma, Inc. (the “Company”) is a clinical-stage biopharmaceutical company discovering and developing novel treatments for autoimmune diseases. The Company has identified Lanthionine Synthetase C-Like 2 (“LANCL2”) as a novel therapeutic target for autoimmune diseases, including inflammatory bowel disease (“IBD”); Crohn’s disease (“CD”), and ulcerative colitis (“UC”). Landos’ wholly-owned lead clinical asset, BT-11, is the first therapeutic that targets LANCL2 and acts locally in the gastrointestinal tract for treatment of inflammatory bowel disease (IBD). The company completed global Phase 2 clinical testing of BT-11 for UC in 2020. Landos is a platform company that continues to discover innovative therapeutic targets (one to two new therapeutic targets per year and their associated drug development programs). Landos also has a robust pipeline of seven product candidates for other autoimmune diseases (lupus, rheumatoid arthritis, multiple sclerosis, type 1 diabetes), several of which Landos anticipates will advance to Phase 1 clinical testing in 2021. Since inception, the Company has devoted substantially all of its resources to performing research and development activities in support of its product development efforts. The Company does not have any products or partnered products approved for sale and has not generated any revenue from commercial product sales. The Company was incorporated in Delaware in January 2017.
Liquidity and capital resources
The Company has incurred net losses and negative cash flows from operations since inception and had an accumulated deficit of $55.7 million and $25.6 million as of December 31, 2020 and 2019, respectively. Since inception through December 31, 2020, the Company has funded operations primarily through the issuance of convertible preferred stock and convertible promissory notes. The Company expects to incur substantial operating losses for at least the next several years and will need to obtain additional financing in order to initiate and complete clinical trials, discover, develop, seek regulatory approvals for and prepare for potential commercialization of its product candidates. There can be no assurance that such financing will be available or will be at terms acceptable to the Company.
As of December 31, 2020, the Company had cash, cash equivalents and marketable securities of $28.1 million, which it believes combined with the proceeds from its initial public offering as discussed in footnote 11 will be sufficient to fund its planned operations for a period of at least 12 months from the date of the issuance of its consolidated financial statements.
2. Summary of significant accounting policies
Basis of presentation
The consolidated financial statements and accompanying notes have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”).
Principals of consolidation
The accompanying consolidated financial statements include the accounts of the Company and its wholly owned subsidiary, Landos Biopharma Australia Pty Ltd. (“Landos Australia”). All intercompany balances and transactions have been eliminated in consolidation.
Use of estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of expenses during the reporting period. Significant estimates and assumptions made in the accompanying consolidated financial statements include but are not limited to accrued liabilities, fair value of equity instruments, and uncertain tax positions. The Company evaluates its estimates and assumptions on an ongoing basis using historical experience and other factors and adjusts those estimates and assumptions when facts and circumstances dictate. Actual results could differ from those estimates.
F-7
COVID-19
In March 2020, the World Health Organization declared the outbreak of the novel coronavirus disease (“COVID-19”) as a pandemic, and the Company expects its operations in all locations to be affected as the virus continues to proliferate. The Company has adjusted certain aspects of its operations to protect employees and customers while still meeting customers’ needs for vital technology. The Company will continue to monitor the situation closely and it is possible that further measures will be implemented. In light of the uncertainty as to the severity and duration of the pandemic, the impact on the financial position is uncertain at this time.
Consolidated financial statements in U.S. dollars
The Company’s functional currency is the U.S. dollar (“dollar” or “$”) since the dollar is the currency of the primary economic environment in which the Company has operated and expects to continue to operate in the foreseeable future. Transactions and balances denominated in dollars are presented at their original amounts. Transactions and balances denominated in foreign currencies have been re-measured to dollars. All transaction gains and losses from re-measurement of monetary balance sheet items denominated in non-dollar currencies are reflected in the consolidated statement of operations and comprehensive loss as other income, net. Net foreign currency transaction losses were not material for the years ended December 31, 2020 and 2019.
Cash and cash equivalents
The Company considers all highly liquid investments purchased with original maturities of three months or less from the purchase date to be cash equivalents. Cash equivalents consist primarily of amounts invested in money market funds and commercial paper and are stated at fair value.
Marketable securities
The Company’s investments in marketable securities are maintained by investment managers and consist of corporate debt securities with original maturities of over 90 days, all of which are considered available-for-sale debt securities. The Company classifies its available-for-sale securities as short-term marketable securities on the consolidated balance sheets, even though the stated maturity date may be one year or more beyond the current consolidated balance sheet date, as the Company views those securities as available for use in current operations, if needed.
Available-for-sale securities are carried at fair value with their unrealized gains and losses included in accumulated other comprehensive loss within stockholders’ (deficit) equity, until such gains and losses are realized in other income (expense), net, within the consolidated statements of operations and comprehensive loss or until an unrealized loss is considered other-than-temporary.
The Company evaluates its investments with unrealized losses for other-than-temporary impairment. When assessing investments for other-than-temporary declines in value, the Company considers such factors as, among other things, how significant the decline in value is as a percentage of the original cost, how long the market value of the investment has been less than its original cost, the Company’s ability and intent to retain the investment for a period of time sufficient to allow for any anticipated recovery in fair value and market conditions. If the Company determines from this analysis that it does not expect to receive cash flows sufficient to recover the entire amortized cost of the security, a credit loss exists, the impairment is considered other-than-temporary and is recognized in the consolidated statements of operations and comprehensive loss.
Concentrations of credit risk
Financial instruments that potentially subject the Company to a concentration of credit risk consist primarily of cash, cash equivalents, and marketable securities. Bank deposits are held by accredited financial institutions and these deposits may at times be in excess of insured limits. The Company limits its credit risk associated with cash and cash equivalents by placing them with financial institutions it believes are of high quality. The Company has not experienced any losses on its deposits of cash or cash equivalents. The Company’s available-for-sale investments primarily consist of high-grade corporate debt, and potentially subject the Company to concentrations of credit risk. The Company has adopted investment guidelines that limit the amounts the Company may invest in any one type of investment and requires all investments held by the Company to be highly rated, thereby reducing credit risk exposure.
F-8
Property and equipment, net
Property and equipment are stated at cost, less accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful lives of the related assets. The estimated useful lives of laboratory equipment, furniture and fixtures ranges from five to seven years. Maintenance, repair and calibration costs are expensed as incurred.
Impairment of long-lived assets
The Company periodically evaluates its long-lived assets, including property and equipment, for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets or group of assets may not be fully recoverable. If indicators of impairment exist and the undiscounted future cash flows that the assets are expected to generate are less than the carrying value of the assets, the Company reduces the carrying amount of the assets through an impairment charge, to their estimated fair values based on a discounted cash flow approach or, when available and appropriate, to comparable market values. In 2020 and 2019, there were no such indicators.
Deferred offering costs
The Company has deferred offering costs consisting of direct legal, accounting, filing and other fees and costs directly attributable to the Company’s planned IPO. The deferred offering costs will be offset against the proceeds received upon the closing of the planned IPO. In the event the planned IPO is terminated, all of the deferred offering costs will be expensed within the Company’s consolidated statement of operations. As of December 31, 2020 and 2019, $1.4 million and $11 thousand of deferred offering costs were capitalized, which are included in current assets on the consolidated balance sheet.
Research and development expenses
Research and development expenses consist primarily of costs incurred for the development of the Company’s lead clinical product candidates BT-11, NX-13 and other pipeline therapeutic assets. Research and development costs consist primarily of external costs related to clinical development, contract manufacturing and discovery as well as personnel costs. Personnel costs consist of salaries and employee benefits. The Company estimates preclinical and clinical study and research expenses based on the services performed, pursuant to contracts with research institutions that conduct and manage preclinical and clinical studies and research services on its behalf. The Company records the costs of research and development activities based upon the estimated amount of services provided but not yet invoiced and includes these costs in accrued liabilities in the consolidated balance sheets. These costs are a component of the Company’s research and development expenses. The Company accrues for these costs based on factors such as estimates of the work completed and in accordance with agreements established with its third-party service providers under the service agreements. The Company makes significant judgments and estimates in determining the accrued liabilities balance in each reporting period. As actual costs become known, the Company adjusts its accrued liabilities. The Company has not experienced any material differences between accrued costs and actual costs incurred.
Share-Based Compensation
The Company accounts for share-based compensation awards in accordance with FASB ASC Topic 718, Compensation—Stock Compensation (ASC 718). ASC 718 requires all share-based payments, including grants of stock options, to be recognized in the consolidated statements of operations and comprehensive loss based on their respective fair values.
The fair value of the Company’s stock options has been determined using the Black-Scholes option-pricing model. The Black-Sholes option-pricing model requires the input of subjective assumptions, including (i) the expected stock price volatility, (ii) the expected term of the award, (iii) the risk-free interest rate and (iv) expected dividends. Due to the lack of historical and implied volatility data of the Company’s common stock, the expected stock price volatility has been estimated based on the historical volatilities of a specified group of companies in Landos’ industry for a period equal to the expected life of the option. The Company selected companies with comparable characteristics, including enterprise value, risk profiles and position within the industry and with historical share price information sufficient to meet the expected term of the stock options. The historical volatility data has been computed using the daily closing prices for the selected companies.
F-9
The expected life of the options granted represents the period of time that options granted are expected to be outstanding and is calculated using the simplified method, which is the mid-point between the vesting date and the end of the contractual term for each option. The risk-free interest rate is based on a zero coupon, United States Treasury instrument whose term is consistent with the expected life of the stock option. The Company has not paid, and does not anticipate paying, cash dividends on its shares of common stock; therefore, the expected dividend yield is zero.
The Company recognizes the grant-date fair value of an award as compensation expense on a straight-line basis over the requisite service period, which typically corresponds to the vesting period for the award. In certain circumstances the amount of compensation cost recognized is adjusted to be at least equal to the portion of the grant-date value of the award that was vested at the balance sheet date. The Company elects to account for forfeitures as they occur and, upon forfeiture of an award prior to vesting, the Company reverses any previously recognized compensation expense related to that award.
Income taxes
Landos Biopharma, Inc. began operations in 2017 and is considered a Subchapter C Corporation and is subject to an entity-level income tax. Income taxes are provided for the tax effects of transactions reported in the financial statements and consist of taxes currently due plus deferred income taxes. Deferred income taxes arise from temporary differences between the tax bases of assets and liabilities and their reported amounts in the financial statements. Deferred income tax assets are also recognized for loss carry forwards available to offset future taxable income. The deferred tax assets and liabilities represent the future tax return consequences of those differences, which will be taxable or deductible when the related assets are recovered or the liabilities are settled. A valuation allowance is established for deferred tax assets if it is considered more likely than not that the asset will not be realized.
In accordance with ASC740-10, Accounting for Income Taxes, (“ASC 740-10”), a company must recognize a tax benefit or expense from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The Company had an unrecorded tax benefit of $0.14 million and $1.1 million due to uncertain tax positions as of December 31, 2020 and 2019, respectively. The Company’s policy for recording interest and penalties is to record them as a component of interest expense and operating expenses, respectively. As of December 31, 2020 and 2019, the Company had no accrued interest or penalties related to uncertain tax positions. The total unrecorded benefit would affect the effective tax rate but for the Company’s valuation allowance. The Company does not expect a material change in unrecognized tax benefits within the next 12 months.
Basic and diluted net loss per share
Basic loss per share is computed by dividing the net loss by the weighted-average number of shares of common stock outstanding during the period. Diluted loss per share is computed by dividing the net loss by the weighted-average number of shares of common stock together with the number of additional shares of common stock that would have been outstanding if all potentially dilutive shares of common stock had been issued. Since the Company was in a loss position for the periods presented, basic net loss per share is the same as diluted net loss per share since the effects of potentially dilutive securities are antidilutive.
Comprehensive loss
The Company’s comprehensive loss is currently comprised of changes in unrealized losses on available-for-sale securities.
Segment reporting
The Company has one operating segment, which is the business of developing and commercializing novel therapeutics for autoimmune diseases. The Company’s chief operating decision maker, its Chief Executive Officer, manages the Company’s operations on an aggregated basis for the purposes of allocating resources and evaluating financial performance.
F-10
Emerging growth company status
The Company is an emerging growth company (“EGC”), as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”). Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies. The Company has elected to use this extended transition period for complying with new or revised accounting standards that have different effective dates for public and private companies until the earlier of the date that it (i) is no longer an EGC or (ii) affirmatively and irrevocably opts out of the extended transition period provided in the JOBS Act. As a result, these combined and consolidated financial statements may not be comparable to companies that comply with the new or revised accounting pronouncements as of public company effective dates.
Recently issued accounting pronouncements not yet adopted
In February 2016, the FASB issued ASU 2016-02—Leases (Topic 842), requiring the recognition of lease assets and liabilities on the balance sheet. The standard: (a) clarifies the definition of a lease; (b) requires a dual approach to lease classification similar to current lease classifications; and (c) causes lessees to recognize leases on the balance sheet as a lease liability with a corresponding right-of-use asset for leases with a lease-term of more than twelve months. The standard is effective for public entities for fiscal years beginning after December 15, 2018 and was initially effective for nonpublic entities for fiscal years beginning after December 15, 2019. In October 2019, the FASB approved a one-year delay in the effective date for non-public companies and, in June 2020, approved an additional one-year delay in the effective date for non-public companies. As a result, the standard is now effective for fiscal years beginning after December 15, 2021. The Company does not expect the adoption of this ASU to have a material impact on its consolidated financial statements.
In June 2016, the FASB issued ASU 2016-13—Financial Instruments (Topic 326) Measurement of Credit Losses on Financial Instrument (“CECL”), which requires an allowance for expected credit losses on financial assets be recognized as early as day one of the instrument. This ASU departs from the incurred loss model which means the probability threshold is removed. It considers more forward-looking information and requires the entity to estimate its credit losses as far as it can reasonably estimate. The ASU is effective for fiscal years beginning after December 15, 2019, including interim periods within those fiscal years, for public business entities that are U.S. Securities and Exchange Commission (SEC) filers, excluding entities eligible to be smaller reporting companies (SRC). For all other public business entities, including SRC, the ASU is effective for fiscal years beginning after December 15, 2022, including interim periods within those fiscal years. Early adoption is permitted for fiscal years beginning after December 15, 2018. The Company elects to adopt the new standard in the annual reporting period beginning after December 31, 2022 and does not expect the adoption of this ASU to have a material impact on the consolidated financial statements.
In August 2018, the FASB issued ASU 2018-15, Intangibles — Goodwill and Other — Internal-Use Software (Subtopic 350-40): Customer’s Accounting for Implementation Costs Incurred in a Cloud Computing Arrangement That Is a Service Contract (“ASU 2018-15”). The intent of this pronouncement is to align the requirements for capitalizing implementation costs incurred in a cloud computing arrangement that is a service contract with the requirements for capitalizing implementation costs incurred to develop or obtain internal-use software as defined in ASC 350-40. Under ASU 2018-15, the capitalized implementation costs related to a cloud computing arrangement will be amortized over the term of the arrangement and all capitalized implementation amounts will be required to be presented in the same line items of the financial statements as the related hosting fees. ASU 2018-15 is effective for fiscal years beginning after December 15, 2019, and interim periods within those fiscal years, with early adoption permitted. The Company adopted ASU 2018-15 for its fiscal year beginning January 1, 2020, and the adoption did not have a material impact on the consolidated financial statements.
3. Fair value measurement
Financial assets and liabilities are recorded at fair value on a recurring basis in the consolidated balance sheet. The carrying values of the Company’s financial assets and liabilities, including cash and cash equivalents, prepaids and other current assets, accounts payable, and accrued expenses approximate their fair value due to the short-term maturity of these instruments. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability (an exit price) in an orderly transaction between market participants at the reporting date. Assets and liabilities recorded at fair value in the consolidated financial statements are categorized based upon the level of judgment associated with the inputs used to measure their fair value. Hierarchical levels are directly related to the amount of subjectivity with the inputs to the valuation of these assets or liabilities as follows:
Level 1—Observable inputs such as unadjusted, quoted prices in active markets for identical assets or liabilities at the measurement date;
F-11
Level 2—Inputs (other than quoted prices included in Level 1) are either directly or indirectly observable inputs for similar assets or liabilities. These include quoted prices for identical or similar assets or liabilities in active markets and quoted prices for identical or similar assets of liabilities in markets that are not active;
Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities
Financial assets and liabilities subject to fair value measurements on a recurring basis and the level of inputs used in such measurements are as follows (in thousands):
|
|
|
December 31, 2020 |
|
|||||||||||||
|
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Aggregate fair value |
|
||||
|
Assets: |
|
|
|
|||||||||||||
|
Money market funds |
|
$ |
265 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
265 |
|
|
Fixed income securities |
|
|
— |
|
|
|
23,343 |
|
|
|
— |
|
|
|
23,343 |
|
|
Asset backed securities |
|
|
— |
|
|
|
2,375 |
|
|
— |
|
|
|
2,375 |
|
|
|
Total assets |
|
$ |
265 |
|
|
$ |
25,718 |
|
|
$ |
— |
|
|
$ |
25,983 |
|
|
|
|
December 31, 2019 |
|
|||||||||||||
|
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Aggregate fair value |
|
||||
|
Assets: |
|
|
|
|||||||||||||
|
Money market funds |
|
$ |
1,333 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
1,333 |
|
|
Commercial paper |
|
— |
|
|
|
3,576 |
|
|
— |
|
|
|
3,576 |
|
||
|
Fixed income securities |
|
— |
|
|
|
27,078 |
|
|
— |
|
|
|
27,078 |
|
||
|
Asset backed securities |
|
— |
|
|
|
13,079 |
|
|
— |
|
|
|
13,079 |
|
||
|
Total assets |
|
$ |
1,333 |
|
|
$ |
43,733 |
|
|
$ |
— |
|
|
$ |
45,066 |
|
The contractual maturities of available for sale securities of December 31, 2020 and 2019, are as follows (in thousands):
|
|
|
As of December 31, |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
|
|
(in thousands) |
|
|||||
|
Within one year |
|
$ |
20,078 |
|
|
$ |
9,088 |
|
|
Within one to five years |
|
|
5,640 |
|
|
|
34,645 |
|
|
Total contractual maturities |
|
$ |
25,718 |
|
|
$ |
43,733 |
|
The Company’s financial instruments consist of Level 1 and Level 2 assets. The Company values its Level 1 assets based on quoted prices in active markets for identical instruments. Level 1 assets consist primarily of highly liquid money market funds that are included in cash equivalents. The Company values its Level 2 assets consisting of commercial paper, fixed income securities, and asset backed securities with the help of a third-party pricing service using quoted market prices for similar instruments or nonbinding market prices that are corroborated by observable market data. The Company uses such pricing data as the primary input, to which no material adjustments have been made during the periods presented, to make its determination and assessments as to the ultimate valuation of these assets. The fair values of these instruments approximate amortized cost.
F-12
4. Consolidated balance sheet components
Property and equipment, net
Property and equipment, net consists of the following:
|
|
|
As of December 31, |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
|
|
(in thousands) |
|
|||||
|
Laboratory equipment |
|
$ |
468 |
|
|
$ |
463 |
|
|
Furniture and fixtures |
|
194 |
|
|
108 |
|
||
|
Construction in process |
|
131 |
|
|
- |
|
||
|
Total property and equipment |
|
793 |
|
|
571 |
|
||
|
Less: accumulated depreciation |
|
|
(349 |
) |
|
|
(212 |
) |
|
Total property and equipment, net |
|
$ |
444 |
|
|
$ |
359 |
|
Depreciation expense for property and equipment was $137 thousand and $103 thousand for the years ended December 31, 2020 and 2019, respectively. Costs for property and equipment not yet placed into service are capitalized as construction in process, and will be depreciated over the relevant estimated useful life once placed into service.
Accrued liabilities
Accrued liabilities consist of the following:
|
|
|
As of December 31, |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
|
|
(in thousands) |
|
|||||
|
Accrued research and development |
|
$ |
1,046 |
|
|
$ |
846 |
|
|
Accrued selling, general, and administrative |
|
840 |
|
|
55 |
|
||
|
Accrued payroll and employee benefits |
|
53 |
|
|
77 |
|
||
|
Total accrued liabilities |
|
$ |
1,939 |
|
|
$ |
978 |
|
5. Convertible promissory notes
On January 24, 2019, the Company entered into a Note Purchase Agreement with Perceptive Sciences Master Fund Ltd. and Perceptive Xontogeny Venture Fund, LP (collectively the “Holders”), for the issuance of convertible promissory notes (the “Notes”) for an aggregate principal amount of $8.0 million, net of issuance costs of $20 thousand. The Company promised to pay the principal amount, together with guaranteed interest at the annual rate of 6%, with principal and accrued interest on the Notes due and payable on January 24, 2020 (unless converted under terms and provisions as set forth within the Agreement).
In the event of a change of control occurring before the maturity date, the holders could elect to (a) convert the outstanding principal and accrued interest into Series A shares at a conversion price equal $3.10 or (b) require the Company to repurchase the Note at a repurchase price equal to 150% of the outstanding principal and accrued interest, in cash. If the Notes have not been paid or converted into equity interests as of January 24, 2020, then each holder could elect, in lieu of cash payment, to convert the outstanding balance into Series A Convertible Preferred Stock at a conversion price equal to $3.10.
The indentures governing the Notes contain covenants that limit the Company’s ability to merge or consolidate and provide for customary events of default, which include nonpayment of principal or interest, breach of covenants, payment defaults, or acceleration of other indebtedness and certain events of bankruptcy. Further, the Notes contained certain clauses related to conversion upon an initial public offering (“IPO”) of the Company or an equity financing.
The Company evaluated the accounting for the Notes and identified an embedded derivative related to repayment upon a change of control, which it evaluated and deemed the likelihood of such an event to be remote. The Company also recorded a beneficial conversion feature discount of $20 thousand in relation to the Notes.
In August 2019, outstanding principal and accrued interest of $8.3 million was converted into 1,101,488 shares of Series B convertible preferred stock at a price of $7.4981 per share upon the closing of the Series B financing.
F-13
6. Convertible preferred stock and stockholders’ deficit
Convertible preferred stock
In August 2019, the Company entered into the Series B Convertible Preferred Stock Purchase Agreement, pursuant to which the Company issued 8,036,574 shares of Series B convertible preferred stock at a price of $7.4981 per share in exchange for proceeds of approximately $51.7 million, net of issuance costs of $0.3 million, and the conversion of $8.3 million in outstanding principal and accrued interest due on the Notes.
The Company’s authorized, issued and outstanding shares, carrying value and aggregate liquidation preferences of its convertible preferred stock are as follows (in thousands, except for share amounts):
|
|
|
As of December 31, 2020 |
|
|||||||||||||
|
Convertible preferred stock |
|
Shares authorized |
|
|
Shares issued and outstanding |
|
|
Carrying value |
|
|
Liquidation preference |
|
||||
|
Series A |
|
|
3,224,034 |
|
|
|
3,224,034 |
|
|
$ |
13,090 |
|
|
$ |
9,995 |
|
|
Series B |
|
|
8,036,574 |
|
|
|
8,036,574 |
|
|
|
59,947 |
|
|
|
60,259 |
|
|
Total |
|
|
11,260,608 |
|
|
|
11,260,608 |
|
|
$ |
73,037 |
|
|
$ |
70,254 |
|
|
|
|
As of December 31, 2019 |
|
|||||||||||||
|
Convertible preferred stock |
|
Shares authorized |
|
|
Shares issued and outstanding |
|
|
Carrying value |
|
|
Liquidation preference |
|
||||
|
Series A |
|
|
3,224,034 |
|
|
|
3,224,034 |
|
|
$ |
13,090 |
|
|
$ |
9,995 |
|
|
Series B |
|
|
8,036,574 |
|
|
|
8,036,574 |
|
|
|
59,947 |
|
|
|
60,259 |
|
|
Total |
|
|
11,260,608 |
|
|
|
11,260,608 |
|
|
$ |
73,037 |
|
|
$ |
70,254 |
|
The characteristics of the Series A and Series B convertible preferred stock are as follows:
Dividends
The holders of convertible preferred stock are entitled to receive noncumulative dividends at a rate per annum equal to 8% of the original issuance price per share, which is $3.10 for Series A convertible preferred stock and $7.4981 for Series B convertible preferred stock, subject to adjustment, if and when declared by the board of directors. These dividends are to be paid in advance of any distributions to common stockholders. No dividends have been declared as of December 31, 2020 and 2019.
Conversion rights
Series A and Series B convertible preferred stock is convertible into common stock at a rate of one share of common stock for each share of convertible preferred stock, subject to adjustment for certain dilutive stock issuance and stock splits.
Optional conversion. Holders of Series A and Series B convertible preferred stock may convert their stock at any time and at the option of the holder.
Automatic conversion. The shares of convertible preferred stock will automatically be converted to shares of common stock upon (a) the closing of a firm commitment underwritten public offering pursuant to an effective registration statement under the Securities Act of 1933, as amended, resulting in at least $50.0 million of gross proceeds and a price of at least 1.5x the Series B original issue price per share; or (b) the vote of at least 66 2/3% of the then outstanding shares of convertible preferred stock voting together as a single class.
Voting rights
Each share of convertible preferred stock has voting rights equal to the number of common shares into which it is convertible. The holders of convertible preferred stock, voting together as a single class, are entitled to elect three directors of the Company. The holders of Series B convertible preferred stock have the right to elect one member of the Company’s Board of Directors. The holders of Series A convertible preferred stock have the right to elect two members of the Company’s Board of Directors. These rights fell away at the time of the IPO as a subsequent event.
F-14
Liquidation preference
A liquidation may be deemed to be occasioned by or to include (i) a consolidation or merger of the Company with or into any other corporation in which the Company’s stockholders of record as constituted immediately prior to such transaction will, immediately after such transaction, fail to hold at least a majority of the voting power of the result of the surviving corporation; or (ii) a sale, lease, transfer, exclusive license or other disposition of all or substantially all of the assets of the Company.
In the event of any voluntary or involuntary liquidation, dissolution or winding up of the Company, or other deemed liquidation event the holders of Series B convertible preferred stock are entitled, before any distribution or payment is made to the holders of Series A convertible preferred stock or common stock, to receive payment based on the original issue price of Series B convertible preferred stock per share, plus all noncumulative unpaid dividends. If upon liquidation, the assets to be distributed to the holders of Series B convertible preferred stock are insufficient to permit payment of the full amounts distributable, the entire assets of the Company shall be distributed ratably among the holders of Series B convertible preferred stock.
After the payment in full of the preferred liquidation preference to the holders of Series B convertible preferred stock, the holders of Series A convertible preferred stock are entitled, before any distribution or payment is made to the holders of common stock, to receive payment based on the original issue price of Series A convertible preferred stock per share, plus all noncumulative unpaid dividends. If upon liquidation, the assets to be distributed to the holders of Series A convertible preferred stock are insufficient to permit payment of the full amounts distributable, the remaining assets of the Company, after such required distributions to the holders of Series B convertible preferred stock, shall be distributed ratably among the holders of Series A convertible preferred stock.
After the payment of the convertible preferred stock liquidation preference, the holders of convertible preferred stock are also entitled to share any remaining available funds on a pro-rata basis with holders of common stock.
Prior to the issuance of the Series B convertible preferred stock, in the event of any voluntary or involuntary liquidation, dissolution or winding up of the Company, or other deemed liquidation event the holders of Series A convertible preferred stock are entitled, before any distribution or payment is made to the holders of common stock, to receive payment based on the original issue price of Series A convertible preferred stock per share, plus all noncumulative unpaid dividends. If upon liquidation, the assets to be distributed to the holders of Series A convertible preferred stock are insufficient to permit payment of the full amounts distributable, the entire assets of the Company shall be distributed ratably among the holders of Series A convertible preferred stock. After the payment of the Series A convertible preferred stock liquidation preference, the holders of convertible preferred stock are also entitled to share any remaining available funds on a pro-rata basis with holders of common stock.
These rights fell away at the time of the IPO as a subsequent event.
Classification
As of December 31, 2020 and 2019, the convertible preferred stock is classified outside of stockholders’ equity (deficit) on the consolidated balance sheet, as mezzanine equity, as events triggering the liquidation preferences are not solely within the Company’s control. Upon the occurrence of certain change in control events that are outside the Company’s control, including a deemed liquidation event, holders of the convertible preferred stock can cause redemption for cash. The carrying values of the convertible preferred stock are adjusted to their liquidation preferences when and if it becomes probable that such an event will occur.
Common stock
Pursuant to the Amended and Restated Certificate of Incorporation effective August 9, 2019, the Company is authorized to issue a total of 37,410,450 shares of common stock, 12,767,909 of which are outstanding as of December 31, 2020.
Holders of common stock are entitled to one vote per share on all matters to be voted on by the stockholders of the Company. Subject to the preferences that may be applicable to any outstanding shares of the convertible preferred stock, the holders of the common stock are entitled to receive ratably such dividends, if any, as may be declared by the Board. No dividends have been declared to date.
F-15
At December 31, 2020 and 2019, the Company has reserved the following shares of common stock for the following purposes:
|
|
|
As of December 31, |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
Convertible preferred stock on an as-converted basis |
|
|
20,549,478 |
|
|
|
20,549,478 |
|
|
Options and restricted stock available for future grants |
|
|
2,003,587 |
|
|
|
3,657,019 |
|
|
Total common stock reserved for issuance |
|
|
22,553,065 |
|
|
|
24,206,497 |
|
Restricted stock
The Company entered into a Strategy & Business Advisor Agreement (“the Agreement”) with Xontogeny LLC (“Xontogeny”), which is a related party due to significant ownership of the Company’s capital stock, on April 24, 2017, which provides for the grant of 3,090,924 shares of common stock to Xontogeny in exchange for the provision of advisory services for a three-year period. The award contains a share repurchase feature at the nominal original share price that is exercisable only if Xontogeny ceases providing the advisory services. The Company accounted for this feature as a forfeiture provision. The shares of common stock vest over an approximate three-year period, 25% of the shares vested upon closing of the Series A financing (the “Series A Closing”), 25% on the first anniversary of the Series A Closing and the remaining 50% vesting monthly over two years commencing on such first anniversary. For accounting purposes, unvested restricted stock awards are not considered issued and outstanding and therefore are not reflected as issued and outstanding in the accompanying statements of convertible preferred stock and stockholders’ deficit until the awards vest.
As of December 31, 2020, the shares were fully vested. As of December 31, 2019, there were 579,548 unvested shares. The shares vested ratably over the service period through September 2020. The Company has determined that the issuance of these shares of common stock to Xontogeny represents compensation for services to be provided under the Agreement. Accordingly, the shares are accounted for similar to a stock award granted to a non-employee of the Company. During the years ended December 31, 2020 and 2019, the Company recognized non-cash compensation expense of $39 thousand and $51 thousand, respectively, in general and administrative expense, related to the vesting of the shares of common stock.
7. Share-based compensation
2019 Equity Incentive Plan
In December 2019, the board of directors of the Company (the “Board”) adopted the 2019 Equity Incentive Plan (the “2019 Plan”). The 2019 Plan provides for the grant of share-based awards, including stock options and restricted stock units, to employees, directors, and non-employee service providers of the Company. In December 2019, the Board authorized 3,657,019 shares for future issuance under the 2019 Plan. All such shares authorized for issuance under the 2019 Plan have been reserved. As of December 31, 2019, the Company had not granted any share-based payment awards; however, during 2020, stock options were awarded to key employees. As of December 31, 2020, there are approximately 2,003,587 shares available for future grants.
A summary of the Company’s stock option activity is as follows:
|
|
|
Number of Shares |
|
|
Weighted Average Exercise Price |
|
|
Weighted Remaining Contract Term (in years) |
|
|
Aggregate Intrinsic Value (in thousands) |
|
||||
|
Balances as of December 31, 2019 |
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
|
— |
|
|
Granted |
|
|
1,653,432 |
|
|
|
1.86 |
|
|
|
|
|
|
|
|
|
|
Exercised |
|
|
(404,214 |
) |
|
|
1.86 |
|
|
|
|
|
|
|
|
|
|
Forfeited |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
Balances as of December 31, 2020 |
|
|
1,249,218 |
|
|
$ |
1.86 |
|
|
|
9.80 |
|
|
$ |
1,225 |
|
|
Exercisable and vested at December 31, 2020 |
|
|
9,142 |
|
|
$ |
1.86 |
|
|
|
9.80 |
|
|
$ |
9 |
|
The total intrinsic value of options exercised was $396 thousand for the year ended December 31, 2020.
F-16
The weighted average fair value of options to purchase common stock granted was $1.01 in the year ended December 31, 2020.
The fair value of each stock option award is estimated on the grant-date using the Black-Scholes option pricing model. The inputs used below are subjective and require significant judgment to determine.
|
|
|
Year Ended December 31, 2020 |
|
|
|
Expected term (in years) |
|
|
5.4 |
|
|
Risk-free interest rate |
|
|
0.32 |
% |
|
Expected volatility |
|
|
63.5 |
% |
|
Dividend rate |
|
|
— |
% |
The following table summarizes stock-based compensation expense for employees, which was included in the statements of operations and comprehensive loss as follows (in thousands):
|
|
|
Year Ended December 31, 2020 |
|
|
|
Research and development |
|
$ |
600 |
|
|
General and administrative |
|
233 |
|
|
|
Total stock-based compensation expense |
|
$ |
833 |
|
At December 31, 2020, the total compensation cost related to unvested stock-based awards granted to employees under the Company’s stock option plan but not yet recognized was approximately $843 thousand. This cost will be amortized on a straight-line basis over the remaining vesting period. The weighted-average remaining recognition period is approximately 1.7 years.
Early Exercise of Employee Options
The terms of the 2019 Plan permit certain option holders to exercise options before their options are vested. The shares of common stock granted upon early exercise that have not vested are subject to repurchase by the Company in the event of termination of the purchaser’s employment, at the price paid by the purchaser. While such shares have been issued, they are not considered outstanding for accounting purposes until they vest and are therefore excluded from shares used in determining loss per share until the repurchase right lapses and the shares are no longer subject to the repurchase feature. The liability is reclassified into common stock and additional paid-in capital as the shares vest and the repurchase right lapses. Accordingly, the Company has recorded the unvested portion of the early exercise proceeds of $765 thousand as a liability in the accompanying balance sheets as of December 31, 2020. As of December 31, 2020, the Company recorded $489 thousand in other current liabilities and $276 thousand in other liabilities related to shares that were subject to repurchase.
8. Commitments and contingencies
Operating leases
The Company leases office space for its corporate headquarters located in Blacksburg, Virginia, under non-cancelable operating leases. The leases expire in 2021. Rent expense is recognized on a straight-line basis over the term of the leases and accordingly, the Company records any differences between cash rent payments and the recognition of rent expense as a deferred rent liability.
Future minimum lease payments as of December 31, 2020, are as follows (in thousands):
|
Year ending December 31: |
|
|
|
|
|
2021 |
|
$ |
108 |
|
|
Total future minimum annual payments |
|
$ |
108 |
|
Rent expense was $229 thousand and $142 thousand for the year ended December 31, 2020 and 2019, respectively.
F-17
Other
Liabilities for loss contingencies arising from claims, assessments, litigation, fines, and penalties and other sources are recorded when it is probable that a liability has been incurred and the amount can be reasonably estimated. The Company believes there is no litigation pending or loss contingencies that could have, either individually or in the aggregate, a material impact on the Company’s financial statements.
9. Income taxes
The following table presents a reconciliation of the statutory federal rate and the Company’s effective tax rate:
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
Federal statutory income tax rate |
|
|
21.00 |
% |
|
|
21.00 |
% |
|
State taxes, net of federal benefit |
|
|
4.19 |
% |
|
|
4.23 |
% |
|
Permanent differences |
|
(0.33 |
)% |
|
|
(2.07 |
)% |
|
|
Other credits |
|
0.23 |
% |
|
4.93 |
% |
||
|
True-ups |
|
(0.01 |
)% |
|
0.02 |
% |
||
|
Foreign rate differential |
|
0.45 |
% |
|
0.06 |
% |
||
|
Change in valuation allowance |
|
|
(25.53 |
)% |
|
|
(28.17 |
)% |
|
Provision for income taxes |
|
|
— |
% |
|
|
— |
% |
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The following table presents the significant components of the Company’s deferred tax assets and liabilities for the periods presented:
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
|
|
(in thousands) |
|
|||||
|
Deferred tax assets/(liabilities): |
|
|
|
|
|
|
|
|
|
Net operating loss carryforwards |
|
$ |
12,294 |
|
|
$ |
4,152 |
|
|
Research and development credit, net of FIN 48 |
|
717 |
|
|
|
1,144 |
|
|
|
Accruals |
|
— |
|
|
224 |
|
||
|
Intangible assets |
|
242 |
|
|
77 |
|
||
|
Unrealized gain |
|
81 |
|
|
74 |
|
||
|
Fixed assets |
|
|
(20 |
) |
|
|
(26 |
) |
|
Valuation Allowance |
|
|
(13,341 |
) |
|
|
(5,645 |
) |
|
Stock Compensation |
|
27 |
|
|
|
— |
|
|
|
Net deferred tax assets |
|
$ |
— |
|
|
$ |
— |
|
At both December 31, 2020, and December 31, 2019, the Company evaluated all significant available positive and negative evidence, including the existence of losses in recent years and management’s forecast of future taxable income, and, as a result, determined it was more likely than not that federal and state deferred tax assets, including benefits related to net operating loss carryforwards, would not be realized. The valuation allowance was increased from $5.6 million at December 31, 2019, to $13.3 million at December 31, 2020.
F-18
As of December 31, 2020, the Company has $43.9 million and $43.9 million of federal and state net operating loss carryforwards. As of December 31, 2019, the Company has $15.4 million and $15.4 million of federal and state net operating loss carryforwards. Federal net operating loss carryforward incurred prior to 2018 as well as the state net operating loss carryforward begin to expire in 2037. Federal net operating losses incurred in 2018 and after have an unlimited carryforward period. The Company also has $0.7 million of Australian net operating loss carryforwards which also have an unlimited carryforward period. Because the Company has incurred cumulative net operating losses since inception, all tax years remain open to examination by U.S. federal, state, and foreign income tax authorities.
The Company had an unrecognized tax benefit of $139 thousand and $1.1 million due to uncertain tax positions as of December 31, 2020 and 2019,, respectively. The Company’s policy for recording interest and penalties is to record them as a component of interest expense and operating expenses, respectively. As of December 31, 2020, the Company had no accrued interest or penalties related to uncertain tax positions. The total unrecorded benefit would affect the effective tax rate but for the Company’s valuation allowance. The Company does not expect a material change in unrecognized tax benefits within the next 12 months.
A reconciliation of the beginning and ending amounts of unrecognized tax benefits is as follows:
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
|
|
(In thousands) |
|
|||||
|
Balance at the beginning of the year |
|
$ |
1,144 |
|
|
$ |
479 |
|
|
Additions for tax positions taken in the current year |
|
|
70 |
|
|
|
666 |
|
|
Reduction for prior tax positions |
|
(1,075 |
) |
|
|
— |
|
|
|
Balance at the end of the year |
|
$ |
139 |
|
|
$ |
1,144 |
|
Potential 382 limitation
The Company’s ability to utilize its net operating loss (“NOL”) and research and development (“R&D”) credit carryforwards may be substantially limited due to ownership changes that may have occurred or that could occur in the future, as required by Section 382 of the Internal Revenue Code of 1986, as amended (the “Code”), as well as similar state provisions. These ownership changes may limit the amount of NOL and R&D credit carryforwards that can be utilized annually to offset future taxable income and tax, respectively. In general, an “ownership change,” as defined by Section 382 of the Code, results from a transaction or series of transactions over a three-year period resulting in an ownership change of more than 50 percent of the outstanding stock of a company by certain stockholders or public groups.
The Company has not completed a study to assess whether one or more ownership changes have occurred since the Company became a loss corporation under the definition of Section 382. If the Company has experienced an ownership change, utilization of the NOL or R&D credit carryforwards would be subject to an annual limitation, which is determined by first multiplying the value of the Company’s stock at the time of the ownership change by the applicable long-term, tax-exempt rate, and then could be subject to additional adjustments, as required. Any such limitation may result in the expiration of a portion of the NOL or R&D credit carryforwards before utilization. Until a study is completed and any limitation known, no amounts are being considered as an uncertain tax position or disclosed as an unrecognized tax benefit under ASC-740. Any carryforwards that expire prior to utilization as a result of such limitations will be removed from deferred tax assets with a corresponding reduction of the valuation allowance. Due to the existence of the valuation allowance, it is not expected that any possible limitation will have an impact on the results of operations of the Company.
On March 27, 2020, the President of the United States signed into law the “Coronavirus Aid, Relief, and Economic Security (“CARES”) Act.” The CARES Act, among other things, includes provisions relating to refundable payroll tax credits, deferment of employer side social security payments, net operating loss carryback periods, alternative minimum tax credit refunds, modifications to the net interest deduction limitations, increased limitations on qualified charitable contributions, and technical corrections to tax depreciation methods for qualified improvement property. At this time, the Company does not believe the CARES Act has a material impact to its overall tax position but will continue to assess in subsequent periods.
F-19
10. Net loss per share common share
The following table sets forth the computation of basic and diluted net loss per share during the periods presented (in thousands, except share and per share amounts):
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
Numerator: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(30,144 |
) |
|
$ |
(13,467 |
) |
|
Denominator: |
|
|
|
|
|
|
|
|
|
Weighted-average shares of common stock issued and outstanding |
|
|
12,473,485 |
|
|
|
12,363,697 |
|
|
Less: weighted-average unvested common stock subject to repurchase |
|
|
(245,663 |
) |
|
|
(970,148 |
) |
|
Weighted-average common stock outstanding used to calculate net loss per common share, basic and diluted |
|
|
12,227,823 |
|
|
|
11,393,549 |
|
|
Net loss per share of common stock, basic and diluted |
|
$ |
(2.47 |
) |
|
$ |
(1.18 |
) |
The following outstanding shares of potentially dilutive securities have been excluded from diluted net loss per common share for the periods presented, because their inclusion would be anti-dilutive:
|
|
|
As of December 31, |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
Convertible preferred stock on an as-converted basis |
|
|
20,549,478 |
|
|
|
20,549,478 |
|
|
Unvested common stock |
|
|
410,602 |
|
|
|
579,549 |
|
|
Stock options to purchase common stock |
|
|
1,249,218 |
|
|
— |
|
|
|
Total |
|
|
22,209,298 |
|
|
|
21,129,027 |
|
11. Subsequent events
On February 3, 2021, the Company completed its initial public offering (“IPO”) in which it issued and sold 6,250,000 shares of its common stock at a public offering price of $16 per share. The Company received net proceeds of $91.2 million from the IPO, after deducting underwriters’ discounts and commissions of $7 million and offering costs of $1.8 million. Offering costs were initially capitalized and consisted of fees and expenses incurred in connection with the sale of common stock in the IPO, including legal, accounting, printing and other IPO-related costs. Upon completion of the IPO, these offering costs were reclassified to stockholders’ equity and offset against the proceeds from the offering on the balance sheet. Immediately prior to the completion of the IPO, all shares of convertible preferred stock then outstanding were converted into 20,549,478 shares of common stock on a one-to-one basis, $72.8 million of convertible preferred stock was reclassified to additional paid-in-capital and $0.2 of convertible preferred stock was reclassified to common stock on the Company’s balance sheet.
On January 27, 2021, the Company’s Board of Directors approved a 1.8249-for-1 stock split of the Company’s outstanding common shares. On January 29, 2021, the Company amended its Amended and Restated Certificate of Incorporation to effect the stock split. The stock split resulted in an adjustment to the preferred share conversion price to reflect a proportional increase in the number of common shares to be issued upon conversion. The accompanying financial statements and notes to financial statements give retroactive effect to the stock split for all periods presented. Also on January 27, 2021, the Board of Directors adopted the 2021 Employee Stock Purchase Plan (the “2021 ESPP”). The 2021 ESPP provides for the purchase by participating employees of 213,000 shares of Common Stock. All such shares authorized for issuance under the 2021 ESPP have been reserved. The number of shares reserved for issuance will automatically increase on January 1st of each year, for a period of not more than ten years by an amount equal to the lessor of (i) 1% of the total number of shares Common Stock outstanding on December 31st of the preceding calendar year or (ii) such smaller number of shares of Common Stock as the Board of Directors may designate. As of March 30, 2021, no shares have been purchased by participating employees. Therefore, all 213,000 reserved shares remain available for future issuance.
F-20
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
We maintain “disclosure controls and procedures,” as defined in Rule 13a-15(e) and Rule 15d-15(e) under the Exchange Act that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to our management, including our principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure.
Our management, with the participation of our Chief Executive Officer, who also serves as our principal financial officer and our principal accounting officer, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act ), as of the end of the period covered by this Form 10-K. Based on such evaluation, our Chief Executive Officer has concluded that as of December 31, 2020, our disclosure controls and procedures were effective to provide reasonable assurance that the information required to be disclosed by us in this Form 10-K was (a) reported within the time periods specified by SEC rules and regulations, and (b) communicated to our management, including our Chief Executive Officer, to allow timely decisions regarding any required disclosure.
Management's Report on Internal Control Over Financial Reporting
This Annual Report on Form 10-K does not include a report of management’s assessment regarding internal control over financial reporting or an attestation report of our independent registered public accounting firm as allowed by the SEC during the transition period for newly public companies.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting identified in management’s evaluation pursuant to Rules 13a-15(d) or 15d-15(d) of the Exchange Act during the quarter ended December 31, 2020 that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Inherent Limitations on Effectiveness of Internal Controls
In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable, not absolute, assurance of achieving the desired control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs. Our management, including our Chief Executive Officer, believes that our disclosure controls and procedures and internal control over financial reporting are designed to provide reasonable assurance of achieving their objectives and are effective at the reasonable assurance level. However, our management does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent all errors and all fraud.
None.
128
Item 10. Directors, Executive Officers and Corporate Governance.
The names of our directors and executive officers, their ages as of March 26, 2021 and certain other information about them are set forth below. There are no family relationships among any of our directors or executive officers, except between our Chairman, Chief Executive Officer and President Josep Bassaganya-Riera and our Chief Scientific Officer Raquel Hontecillas, who are married.
Executive Officers
Josep Bassaganya-Riera, Ph.D., age 46, has served as our Chairman, President and Chief Executive Officer since our founding in January 2017. Dr. Bassaganya-Riera has served as the Director of the Nutritional Immunology and Molecular Medicine Laboratory since July 2002 and the Chairman of the board of directors of Biotherapeutics Inc. since October 2008. He previously served as the Chief Executive Officer of Biotherapeutics from October 2008 to September 2017 and a Research Professor of Immunology at Virginia Tech from December 2002 to May 2020. Dr. Bassaganya-Riera holds a degree in Veterinary Medicine from Universitat Autònoma de Barcelona and a Ph.D. in Nutrition and Immunology from Iowa State University. Our board of directors believes that Dr. Bassaganya-Riera is qualified to serve as a director based on his role as the founder of our company, his service as our President and Chief Executive Officer and his extensive management and technical experience in the pharmaceutical industry. Dr. Bassaganya-Riera serves on the Nominating and Corporate Governance, Compensation and Audit Committees. We expect that within one year of our listing on Nasdaq, Dr. Bassaganya-Riera will resign from each of these committee and will be replaced by an independent director; at that point, all members of each committee of the Board will be independent.
Raquel Hontecillas, Ph.D., age 49, has served as our Chief Scientific Officer since our founding in January 2017. Dr. Hontecillas is the Co-Director of the Nutritional Immunology and Molecular Medicine Laboratory, a position she has held since July 2002, and the Chief Scientific Officer of Biotherapeutics, Inc., a position she has held since October 2008. She previously served as a Research Professor of Immunology at Virginia Tech from July 2008 to July 2020. Dr. Hontecillas holds a degree in Veterinary Medicine from Universidad Complutense University de Madrid, Spain and a Ph.D. in Immunology from Iowa State University.
Jyoti Chauhan, MS, RAC, age 32, has served as our Executive Director of Global Clinical Operations and Regulatory Affairs since August 2019. She previously served as our Clinical Project Manager from February 2018 to July 2018 and our Program Director, Clinical Operations and Regulatory Affairs from July 2018 to August 2019. Ms. Chauhan previously served as the Manager of Regulatory Affairs at NexImmune, Inc. from March 2018 to August 2018, the Senior Regulatory Affairs Specialist at the International Partnership for Microbicides from September 2017 to March 2018 and the Regulatory Affairs Specialist at Technical Resources International, Inc. from March 2016 to September 2017, and currently serves as a Principal at Nova Technology Inc., a position she has held since October 2015. Ms. Chauhan holds a B.S. in Biotechnology with Chemistry, Botany and Zoology from Hemwati Nandan Bahuguna Garhwal University and a M.S. in Pharmaceutical Sciences from the Narsee Monjee Institute of Management Studies.
Non-Employee Directors
Christopher Garabedian, age 54, has served as a director since September 2017. He founded Xontogeny in June 2016 and serves as its Chairman and Chief Executive Officer. Mr. Garabedian also serves as a Senior Advisor at the Boston Consulting Group, a position he has held since January 2016, and as a Portfolio Manager at Perceptive Advisors, a position he has held since May 2017. He previously served as the President and Chief Executive Officer of Sarpeta Therapeutics, Inc. from January 2011 to March 2015, as well as a member of the board of directors from June 2010 to March 2015. Mr. Garabedian earned a B.S. in Marketing from the University of Maryland. Our board of directors believes that Mr. Garabedian is qualified to serve as a director based on his extensive management experience in the biopharmaceutical industry.
Konstantin Poukalov, age 37, has served as a director since August 2019. Mr. Poukalov has served a Managing Director at Perceptive Advisors since March 2019 and as the Chief Business Officer of ARYA Sciences Acquisition Corp II since June 2020. He previously served as the Chief Financial Officer of Kadmon Holdings, Inc. from July 2014 to October 2018. He has served on the board of directors of Lyra Therapeutics since January 2020. Mr. Poukalov holds a B.S. in Electrical Engineering from Stony Brook University. Our board of directors believes
129
that Mr. Poukalov is qualified to serve as a director based on his extensive financial and industry experience. Mr. Poukalov serves as a member of the Nominating and Corporate Governance, Compensation and Audit Committees. We expect that within one year of our listing on Nasdaq, Mr. Poukalov will resign from the Audit Committee and will be replaced by an independent director; at that point, all members of the Audit Committee will be independent.
Roderick Wong, M.D., age 43, has served as a director since August 2019. Dr. Wong has served as Managing Partner and Chief Investment Officer of RTW Investments, LP since 2010. Dr. Wong previously served as the President, Chief Executive Officer and Chairman of Health Sciences Acquisition Corporation from December 2018 to December 2019. He has served as Chairman of the board of directors of Rocket Pharmaceutics, Inc. since July 2015 and as a member of the board of directors of Avidity Biosciences, Inc. since June 2020. Dr. Wong holds a B.S. in Economics from Duke University, an M.D. from the University of Pennsylvania Medical School and an M.B.A. from Harvard Business School. Our board of directors believes that Dr. Wong is qualified to serve as a director based on his experience as an investor and an executive in the biopharmaceutical industry.
Jean-Frédéric Colombel, M.D., age 64, has served as a director since January 2021. Dr. Colombel currently serves as a Professor of Medicine and Director of the Leona and Harry B. Helmsley Charitable Trust Inflammatory Bowel Disease, Icahn School of Medicine, New York, positions he has held since 2013. Since 2013, Dr. Colombel has also served as the Co-Director of the Susan and Leonard Feinstein Inflammatory Bowel Disease Center, Icahn School of Medicine, New York. Since 2017, Dr. Colombel has served as a member of the Committee on Appointments, Promotions and Tenure at the Icahn School of Medicine, New York. Dr. Colombel has served as an associate editor of Gastroenterology since 2016 and has been a member of the National Scientific Advisory Committee at the Crohn’s & Colitis Foundation of America since 2015. Dr. Colombel holds an HDR (Habilitation a Diriger les Recherches) from Universite Lille II, France, a DEA (Diplome d’Etudes Approfondies) from Universite Paris VII, France, and an M.D. from Université Lille II, France. Our board of directors believes Dr. Colombel is qualified to serve as a director based on his extensive medical experience and expertise. Mr. Colombel serves as a member of the Nominating and Governance, Compensation and Audit Committees.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Securities Exchange Act of 1934, as amended, or the Exchange Act, requires our directors and executive officers, and persons who own more than ten percent of a registered class of our equity securities, to file with the SEC initial reports of ownership and reports of changes in ownership of common stock and other equity securities of the Company. Officers, directors and greater than ten percent stockholders are required by SEC regulation to furnish the Company with copies of all Section 16(a) forms they file.
To our knowledge, based solely on a review of the copies of such reports furnished to us and written representations that no other reports were required, during the fiscal year ended December 31, 2020, all Section 16(a) filing requirements applicable to its officers, directors and greater than ten percent beneficial owners were complied with.
Audit Committee and Audit Committee Financial Expert
The Audit Committee of the board of directors was established by the board in accordance with Section 3(a)(58)(A) of the Exchange Act to oversee the Company’s corporate accounting and financial reporting processes and audits of its financial statements. The Audit Committee is comprised of three directors: Josep Bassaganya-Riera, Jean-Frederic Colombel and Konstantin Poukalov, with Mr. Poukalov serving as chair.
The board of directors reviews the Nasdaq Stock Market, or Nasdaq, listing standards definition of independence for Audit Committee members on an annual basis. Our board of directors has determined that Dr. Colombel meets the independence requirements of the Sarbanes-Oxley Act of 2002, as amended, or the Sarbanes-Oxley Act, Rule 10A-3 under the Securities Exchange Act of 1934, or the Exchange Act, and the applicable listing standards of Nasdaq. We expect that within one year of our listing on Nasdaq, Dr. Bassaganya-Riera and Mr. Poukalov will each resign from the audit committee and will be replaced by an independent director; at that point, all members of the audit committee will be independent. Each member of our audit committee can read and understand fundamental financial statements in accordance with Nasdaq audit committee requirements. In arriving at this determination, the board has examined each audit committee member’s scope of experience and the nature of their prior and/or current employment.
The board of directors has also determined that Mr. Poukalov qualifies as an “audit committee financial expert,” as defined in applicable SEC rules. The board made a qualitative assessment of Mr. Poukalov’s level of knowledge and
130
experience based on a number of factors, including his formal education and previous and current experience in financial and accounting roles.
Stockholder Recommendations for Director Candidates
Historically, we have not provided a formal process related to stockholder communications with the board of directors. Nevertheless, every effort has been made to ensure that the views of stockholders are heard by the board or individual directors, as applicable, and that appropriate responses are provided to stockholders in a timely manner. The Company believes its responsiveness to stockholder communications to the board has been excellent.
Code of Business Conduct and Ethics
We have adopted a Code of Business Conduct and Ethics, which is applicable to all of our employees, executive officers and directors. This includes our principal executive officer, principal financial officer and principal accounting officer or controller, or persons performing similar functions. The full text of the Code of Business Conduct and Ethics is available on our website at www.landosbiopharma.com. We intend to post on our website all disclosures that are required by law or the listing standards of the Nasdaq Stock Market concerning any amendments to, or waivers from, any provision of the Code of Business Conduct and Ethics.
Item 11. Executive Compensation.
This section provides a summary of the compensation of our “named executive officers,” who are the three executive officers listed in the “Summary Compensation Table” below. In addition to presenting quantitative compensation information in the tables below, this section also provides a qualitative description of the material factors helpful to an understanding of such data.
The following table sets forth information regarding compensation awarded to, earned by and paid to our named executive officers with respect to the years ended December 31, 2020 and 2019.
|
Name and principal position |
|
Year |
|
Salary ($)(1) |
|
|
Bonus ($)(2) |
|
|
Option awards ($)(3) |
|
|
Non-equity incentive plan compensation ($)(4) |
|
|
All other compensation ($)(5) |
|
|
Total ($) |
|
||||||
|
Josep Bassaganya-Riera, Ph.D.(6) |
|
2020 |
|
|
502,500 |
|
|
— |
|
|
|
1,020,000 |
|
|
|
226,125 |
|
|
|
13,150 |
|
|
|
1,761,775 |
|
|
|
|
|
2019 |
|
|
382,083 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
|
382,083 |
|
||||
|
Chairman, President, Chief Executive Officer |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Raquel Hontecillas, Ph.D. |
|
2020 |
|
|
172,581 |
|
|
|
50,000 |
|
|
|
680,000 |
|
|
— |
|
|
|
6,177 |
|
|
|
908,758 |
|
|
|
|
|
2019 |
|
|
89,583 |
|
|
|
30,000 |
|
|
— |
|
|
— |
|
|
— |
|
|
|
119,583 |
|
|||
|
Chief Scientific Officer |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Jyoti Chauhan, MS, RAC |
|
2020 |
|
|
224,194 |
|
|
|
50,000 |
|
|
|
680,000 |
|
|
— |
|
|
|
7,226 |
|
|
|
961,420 |
|
|
|
|
|
2019 |
|
|
170,833 |
|
|
|
50,000 |
|
|
— |
|
|
— |
|
|
— |
|
|
|
220,833 |
|
|||
|
Executive Director, Global Clinical Operations and Regulatory Compliance |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
Each named executive officer’s base salary is a fixed component of annual compensation for performing specific duties and functions, and has been established taking into account each individual’s roles, responsibilities, skills and expertise. In 2019, we paid annual base salaries of $350,000, $75,000 and $150,000 to each of Drs. Bassaganya-Riera and Hontecillas and Ms. Chauhan, respectively, which were subsequently increased to $385,000, $100,000 and $200,000, respectively, during 2019 and $502,500, $250,000 and $250,000, respectively, during 2020. |
|
(2) |
Represents discretionary bonuses awarded to Dr. Hontecillas and Ms. Chauhan in 2019 and 2020 pursuant to their terms of their offer letter agreements. See “—Agreements with our named executive officers and potential payments upon termination of employment.” |
|
(3) |
In accordance with SEC rules, the amounts in this column represent the aggregate grant date fair value of the option awards determined in accordance with FASB ASC Topic 718. See “—Outstanding equity awards at December 31, 2020.” For stock option valuations, the fair value of each stock option award and purchase rights under the 2019 Plan are estimated on the date of grant using the Black-Scholes valuation model and the following assumptions: exercise price, expected term, current price, expected volatility, risk-free interest rate and expected dividend yield. The dividend yield reflects that we have not paid any cash dividends since inception and do not intend to pay any cash dividends in the foreseeable future. |
131
|
(4) |
Represents a performance-based bonus awarded based upon the achievement of individual and company performance goals and conditions at our company as determined by our board of directors. See “—Agreements with our named executive officers and potential payments upon termination of employment.” |
|
(5) |
Represents employer contributions to retirement plans. See “—Retirement benefits and other compensation.” |
|
(6) |
Dr. Bassaganya-Riera is also our Chairman, but he did not receive any additional compensation in his capacity as a director in 2019 or 2020. |
Outstanding equity awards at December 31, 2020
The following table sets forth certain information about outstanding equity awards granted to our named executive officers that were outstanding as of December 31, 2020.
|
|
|
Option Awards(1) |
||||||||||
|
Name |
|
Grant Date |
|
Number of Securities Underlying Unexercised Options (#) Exercisable(2) |
|
Number of Securities Underlying Unexercised Options (#) Unexercisable |
|
Option Exercise Price($)(3) |
|
|
Option Expiration Date |
|
|
Josep Bassaganya-Riera, Ph.D. |
|
10/2/2020 |
|
— |
|
410,602 (4) |
|
|
1.86 |
|
|
1/1/2030 |
|
Raquel Hontecillas, Ph.D. |
|
10/20/2020 |
|
— |
|
273,735(4) |
|
|
1.86 |
|
|
1/1/2030 |
|
Jyoti Chauhan, MS, RAC |
|
10/20/2020 |
|
— |
|
273,735(4) |
|
|
1.86 |
|
|
1/1/2030 |
|
(1) |
All of the awards listed in this table were granted under our 2019 Equity Incentive Plan. |
|
(2) |
As of December 31, 2020, Dr. Bassaganya-Riera had exercised the option shown in this table with respect to 136,867 shares of common stock exercisable at December 31, 2020 and Dr. Hontecillas and Ms. Chauhan had exercised the options shown in this table with respect to 91,245 shares of common stock exercisable at December 31, 2020. |
|
(3) |
All of the option awards listed in the table were granted with a per share exercise price equal to or above the estimated fair value of our common stock on the date of grant, as determined in good faith by our board of directors. |
|
(4) |
The shares subject to this award vest and become exercisable over a three-year period commencing on January 1, 2020, with 25% of the option vesting immediately, 25% vesting on January 1, 2021 and the remaining 50% vesting in equal monthly installments over the twenty-four months thereafter. |
Agreements with our named executive officers and potential payments upon termination of employment
We have entered into an employment agreement, effective January 1, 2020, or the Agreement, with Dr. Josep Bassaganya-Riera, that provides for an annual base salary of $502,500, which is subject to increase during the term of the Agreement at the discretion of our board of directors, and an annual discretionary bonus with a target equal to 45% of Dr. Bassaganya-Riera’s annual base salary based upon the achievement of individual and company performance goals and conditions at our company as determined by our board of directors. The initial term of the employment agreement, or the Initial Term, is two years from the effective date of the employment agreement, and following the Initial Term, Dr. Bassaganya-Riera’s employment period will be automatically renewed for successive one-year periods unless either we or Dr. Bassaganya-Riera terminates it or elects not to renew it.
The Agreement further provides for the grant of an option to purchase 547,470 shares of common stock to Dr. Bassaganya-Riera, which was granted on October 2, 2020, at an exercise price of $1.86 per share. The option vested and became exercisable over a three-year period commencing on January 1, 2020, with 25% of the option vesting immediately, 25% vesting on January 1, 2021 and the remaining 50% vesting in equal monthly installments over the twenty-four months thereafter. Pursuant to the Agreement, if, subsequent to our initial public offering and prior to September 1, 2022, our company’s market capitalization, as determined by multiplying our daily volume weighted-average stock price by the number of shares of common stock then outstanding, exceeds $2,000,000,000 over a consecutive 10-calendar day period, or over 15 calendar days within a 30-day calendar period, Dr. Bassaganya-Riera is entitled to receive a further option to purchase 273,735 shares of common stock.
Pursuant to the Agreement, if we terminate Dr. Bassaganya-Riera’s employment without “Cause,” or if Dr. Bassaganya-Riera terminates his employment for “Good Reason” (each, as defined in the Agreement), in exchange for execution and making effective and irrevocable, a general release in a form acceptable to us, compliance with certain non-competition and non-solicitation obligations, resignation from all positions with our company and return of all company property, he will be eligible for payment of his base salary for 12 months, or if during the Initial Term, payment of his base salary for the longer of the remainder of the Initial Term or 12 months, and any then-outstanding equity awards that would have become vested during the 12 months following the termination date will become fully vested and exercisable. Dr. Bassaganya-Riera will also be eligible for COBRA premiums, with the cost of the regular premium for such benefits shared in the same relative proportion by Dr. Bassaganya-Riera and us as in effect on his termination date until the earlier of 12 months or the date Dr. Bassaganya-Riera becomes eligible for health benefits through another employer or otherwise becomes ineligible for COBRA. Such severance and acceleration benefits are conditioned upon Dr. Bassaganya-Riera’s execution of and
132
compliance with an effective and irrevocable general release, compliance with certain non-competition and non-solicitation obligations, resignation from all positions with us and return of all our property.
We intend to enter into employment agreements with Dr. Hontecillas and Ms. Chauhan in the future. We have engaged Dr. Hontecillas and Ms. Chauhan with an offer letter that provides for an initial base salary, discretionary bonus opportunity and equity compensation, as described in “—Summary compensation table.” Neither of our current offer letter agreements with Dr. Hontecillas and Ms. Chauhan includes any severance entitlements or other potential payments in the event of any change in control, termination, or other defined triggering events.
We do not maintain any other offer letters or employment agreements with any other named executive officers.
Retirement benefits and other compensation
Our named executive officers were eligible to participate in our employee benefits, including health insurance and group life insurance benefits, on the same basis as our other employees. We maintain an employee savings plan pursuant to Section 401(k) of the Internal Revenue Code covering all eligible employees. We have elected to make non-elective contributions totaling to 3% of an eligible employee’s gross salary. Our named executive officers received 3% non-elective contributions once the plan became active in 2020. We generally do not provide other perquisites or personal benefits except in limited circumstances, and we did not provide any such perquisites or personal benefits to our named executive officers in 2020.
Non-Employee Director Compensation
Non-Employee Director Compensation Policy
During the year ended December 31, 2020, we did not pay any fees to, make any equity awards or non-equity awards to, or pay any other compensation to the non-employee members of our board of directors for their services as directors. Our non-employee directors only received reimbursement of their actual out-of-pocket costs and expenses incurred in connection with attending board meetings. Dr. Bassaganya-Riera, our President and Chief Executive Officer, is also the Chairman of our board of directors, but did not receive any additional compensation for his service as a director.
In recognition of the increased responsibilities of our directors as directors of a public company, our board of directors has adopted a non-employee director compensation policy, effective March 30, 2021, pursuant to which each of our directors who is not an employee or consultant of our company is eligible to receive compensation for service on our board of directors and committees of our board of directors.
Each eligible director will receive an annual cash retainer of $45,000 for serving on our board of directors, and the independent chairperson of the board of directors will receive an additional annual cash retainer of $30,000 for his or her service. The chairperson of the audit committee of our board of directors will be entitled to additional annual cash retainer of $20,000, the chairperson of the compensation committee of our board of directors will be entitled to additional annual cash retainer of $15,000 and the chairperson of the nominating and corporate governance committee of our board of directors will be entitled to additional annual cash retainer of $10,000. The members of the audit committee will be entitled to an additional annual cash retainer of $10,000, the members of the compensation committee of our board of directors will be entitled to additional annual cash retainer of $7,500 and the members of the nominating and corporate governance committee of our board of directors will be entitled to an additional annual cash retainer of $5,000; however, in each case such cash retainer is payable only to members who are not the chairperson of such committee.
In addition, each new eligible director who joins our board of directors will be granted a non-statutory stock option to purchase 36,000 shares of our common stock under our 2019 Equity Incentive Plan, with the shares vesting in 36 equal monthly installments, subject to continued service as a director through the vesting date.
On the date of each annual meeting of our stockholders, each eligible director who continues to serve as a director of our company following the meeting will be granted a non-statutory stock option to purchase 18,000 shares of our common stock under our 2019 Equity Incentive Plan, with the shares vesting on the the first anniversary of the date of grant, subject to continued service as a director though the applicable vesting date.
The exercise price per share of each stock option granted under the non-employee director compensation policy will be equal to the closing price of our common stock on the Nasdaq Global Market on the date of grant. Each stock
133
option will have a term of ten years from the date of grant, subject to earlier termination in connection with a termination of the eligible director’s continuous service with us.
We intend to adopt a non-employee director compensation policy, pursuant to which our non-employee directors will be eligible to receive compensation for service on our board of directors and committees of our board of directors, in the future.
Compensation Committee Interlocks and Insider Participation
None of our directors who serve as a member of our compensation committee is, or has at any time during the past year been, one of our officers or employees. None of our executive officers currently serves, or in the past year has served, as a member of the board of directors or compensation committee of any other entity that has one or more executive officers serving on our board of directors or compensation committee.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The following table sets forth information regarding beneficial ownership of our capital stock by:
|
|
• |
each person, or group of affiliated persons, known by us to beneficially own more than 5% of our common stock; |
|
|
• |
each of our directors; |
|
|
• |
our named executive officer; and |
|
|
• |
all of our current executive officers and directors as a group. |
We have determined beneficial ownership in accordance with the rules of the SEC. Under these rules, beneficial ownership includes any shares of common stock as to which the individual or entity has sole or shared voting power or investment power. Applicable percentage ownership is based on 40,117.598 shares of common stock outstanding as of March 15, 2021. In computing the number of shares beneficially owned by an individual or entity and the percentage ownership of that person, shares of common stock subject to options held by such person that are currently exercisable or will become exercisable within 60 days of March 15, 2021 are considered outstanding, although these shares are not considered outstanding for purposes of computing the percentage ownership of any other person.
Unless noted otherwise, the address of all listed stockholders is c/o Landos Biopharma, Inc., 1800 Kraft Drive, Suite 216, Blacksburg, Virginia 24060.
134
Except as indicated by the footnotes below, we believe, based on information furnished to us, that each of the stockholders listed has sole voting and investment power with respect to the shares beneficially owned by the stockholder unless noted otherwise, subject to community property laws where applicable.
|
Name of Beneficial Owner |
|
Number of Shares Beneficially Owned |
|
|
Percentage of Shares Beneficially Owned |
|
||
|
Greater than 5% stockholders |
|
|
|
|
|
|
|
|
|
Entities affiliated with Perceptive(1) |
|
|
17,960,839 |
|
|
|
44.77 |
% |
|
Josep Bassaganya-Riera, Ph.D.(2) |
|
|
9,820,242 |
|
|
|
24.48 |
|
|
Entities affiliated with RTW(3) |
|
|
4,275,722 |
|
|
|
10.66 |
|
|
Named Executive Officers and Directors |
|
|
|
|
|
|
|
|
|
Josep Bassaganya-Riera, Ph.D.(2) |
|
|
9,820,242 |
|
|
|
24.48 |
|
|
Raquel Hontecillas, Ph.D.(4) |
|
|
182,490 |
|
|
* |
|
|
|
Jyoti Chauhan, MS, RAC(5) |
|
|
148,273 |
|
|
* |
|
|
|
Christopher Garabedian(6) |
|
|
3,090,924 |
|
|
* |
|
|
|
Konstantin Poukalov(7) |
|
|
17,960,839 |
|
|
|
44.77 |
|
|
Roderick Wong, M.D.(8) |
|
|
4,275,722 |
|
|
|
10.66 |
|
|
Jean-Frederic Colombel, M.D. |
|
|
— |
|
|
* |
|
|
|
All current executive officers and directors as a group (7 persons) |
|
|
35,478,490 |
|
|
|
79.91 |
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
This information has been obtained from a Schedule 13G/A filed on February 18, 2021 by Perceptive Advisors LLC. Consists of (a) 7,299,751 shares of common stock held by Perceptive Life Sciences Master Fund, Ltd. (b) 5,799,564 shares of common stock held by Perceptive Xontogeny Venture Fund, LP, (c) 3,090,924 shares of common stock held by Xontogeny, LLC and (d) 1,770,600 shares of common stock held by PX Venture (A), LLC. Perceptive Life Sciences Master Fund Ltd., Perceptive Advisors LLC and Joseph Edelman have shared voting and dispositive power with respect to the shares held by Perceptive Life Sciences Master Fund Ltd. Perceptive Advisors LLC serves as the investment manager to Perceptive Life Sciences Master Fund Ltd. and may be deemed to beneficially own the securities directly held by Perceptive Life Sciences Master Fund Ltd. Mr. Edelman is the managing member of Perceptive Advisors LLC and may be deemed to beneficially own the securities directly held by Perceptive Life Sciences Master Fund Ltd. The principal address of Perceptive Advisors LLC is 51 Astor Place, 10th Floor New York, NY 10003. |
|
(2) |
Consists of (a) 8,229,881 shares of common stock held by Dr. Bassaganya-Riera, as Trustee of the Josep Bassaganya-Riera Revocable Trust Agreement dated April 9, 2018, as amended, (b) 766,405 shares of common stock held by Dr. Bassaganya-Riera, as Trustee of the Josep Bassaganya-Riera Grantor Retained Annuity Trust No. 1 dated June 28, 2018 and (c) 823,956 shares of common stock held by Dr. Bassaganya-Riera, as Trustee of the Josep Bassaganya-Riera Grantor Retained Annuity Trust No. 2 dated March 26, 2019. Of these shares, 216,706 are subject to continued vesting requirements |
|
(3) |
This information has been obtained from a Schedule 13G filed on February 16, 2021 by RTW Investments, LP. Consists of (a) 2,124,235 shares of common held by RTW Master Fund, Ltd. (b) 556,941 shares of common stock held by RTW Innovation Master Fund, Ltd. and (c) 1,216,907 shares of common stock held by RTW Venture Fund Limited. RTW Investments, LP is the investment manager of each funds and has the power to vote and the power to direct the disposition of all such shares. Roderick Wong is the Managing Partner of RTW Investments, LP. The address of RTW Investments, LP and Roderick Wong is 412 West 15th Street, Floor 9, New York, New York 10011. The address of RTW Master Fund, Ltd. is c/o Intertrust Corporate Services (Cayman) Limited, 190 Elgin Avenue, George Town, Grand Cayman KY1-9001, Cayman Islands. |
135
|
(4) |
Consists of 182,490 shares of common stock held by Dr. Hontecillas, as Trustee of the Raquel Hontecillas-Magarzo Revocable Trust Agreement dated April 9, 2018, as amended and (b) 53,340 shares of common stock issuable to Dr. Hontecillas upon the exercise of outstanding options exercisable within 60 days of March 15, 2021. |
|
(5) |
Consists of (a) 91,245 shares of common stock held by Ms. Chauhan and (b) 144,585 shares of common stock issuable to Ms. Chauhan upon the exercise of outstanding options exercisable within 60 days of March 15, 2021. |
|
(6) |
Consists of 3,090,924 shares of common stock held by Xontogeny LLC. Mr. Garabedian is the Chairman and Chief Executive Officer of Xontogeny and may be deemed to beneficially own these shares. |
|
(8) |
Consists of (a) 2,124,235 shares of common held by RTW Master Fund, Ltd. (b) 556,941 shares of common stock held by RTW Innovation Master Fund, Ltd. and (c) 1,216,907 shares of common stock held by RTW Venture Fund Limited. RTW Investments, LP is the investment manager of each funds and has the power to vote and the power to direct the disposition of all such shares. Dr. Wong is the Managing Partner of RTW Investments, LP. |
Equity Compensation Plan Information
The following table contains certain information with respect to our equity compensation plan in effect as of December 31, 2020.
|
Plan category |
|
Shares of common stock to be issued upon exercise of outstanding options (#) |
|
|
Weighted- average exercise price of outstanding options ($) |
|
|
Number of shares of common stock remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (#) |
|
|||
|
Equity compensation plans approved by stockholders |
|
|
1,249,209 |
|
|
$ |
1.86 |
|
|
|
2,003,587 |
|
|
Equity compensation plans not approved by stockholders |
|
— |
|
|
|
— |
|
|
— |
|
||
|
Total |
|
|
1,249,209 |
|
|
$ |
1.86 |
|
|
|
2,003,587 |
|
|
(1) |
Reflects shares of common stock available for future issuance under our 2019 Equity Incentive Plan at December 31, 2020. |
136
Item 13. Certain Relationships and Related Transactions, and Director Independence.
Related-Person Transactions Policy and Procedures
In February 2021, we adopted a related person transaction policy that sets forth our procedures for the identification, review, consideration and approval or ratification of related person transactions. For purposes of our policy only, a related person transaction is a transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships, in which we and any related person are, were or will be participants in which the amount involved exceeds $120,000. Transactions involving compensation for services provided to us as an employee or director are not covered by this policy. A related person is any executive officer, director or beneficial owner of more than 5% of any class of our voting securities, including any of their immediate family members and any entity owned or controlled by such persons.
Under the policy, if a transaction has been identified as a related person transaction, including any transaction that was not a related person transaction when originally consummated or any transaction that was not initially identified as a related person transaction prior to consummation, our management must present information regarding the related person transaction to our audit committee, or, if audit committee approval would be inappropriate, to another independent body of our board of directors, for review, consideration and approval or ratification. The presentation must include a description of, among other things, the material facts, the interests, direct and indirect, of the related persons, the benefits to us of the transaction and whether the transaction is on terms that are comparable to the terms available to or from, as the case may be, an unrelated third party or to or from employees generally. Under the policy, we will collect information that we deem reasonably necessary from each director, executive officer and, to the extent feasible, significant stockholder to enable us to identify any existing or potential related-person transactions and to effectuate the terms of the policy. In addition, under our Code of Business Conduct and Ethics, our employees and directors have an affirmative responsibility to disclose any transaction or relationship that reasonably could be expected to give rise to a conflict of interest. In considering related person transactions, our audit committee, or other independent body of our board of directors, will take into account the relevant available facts and circumstances including:
|
|
• |
the risks, costs and benefits to us; |
|
|
• |
the impact on a director’s independence in the event that the related person is a director, immediate family member of a director or an entity with which a director is affiliated; |
|
|
• |
the availability of other sources for comparable services or products; and |
|
|
• |
the terms available to or from, as the case may be, unrelated third parties or to or from employees generally. |
The policy requires that, in determining whether to approve, ratify or reject a related person transaction, our audit committee, or other independent body of our board of directors, must consider, in light of known circumstances, whether the transaction is in, or is not inconsistent with, our best interests and those of our stockholders, as our audit committee, or other independent body of our board of directors, determines in the good faith exercise of its discretion.
Private placements of our securities
Transactions with Perceptive
Convertible promissory notes and issuance of Series B preferred stock
In January 2019, we entered into a note purchase agreement with Perceptive Life Sciences Master Fund, Ltd. and Perceptive Xontogeny Venture Fund, LP, pursuant to which we issued convertible promissory notes in an aggregate principal amount of $8.0 million, net of issuance costs of $20,000. The maturity date of the notes was January 24, 2020 and the notes bore interest at a rate of 6%. In August 2019, the holders elected to convert their outstanding convertible promissory notes in the aggregate principal amount of $8.3 million into an aggregate of 1,101,488 shares of Series B redeemable convertible preferred stock.
137
Series B preferred stock financing
In August 2019, we entered into a preferred stock purchase agreement with certain investors, including beneficial owners of greater than 5% of our capital stock, pursuant to which we issued and sold to such investors an aggregate of 8,036,574 shares of Series B convertible preferred stock at a purchase price of $7.4981 per share for an aggregate purchase price of $51.7 million.
The following table sets forth the aggregate number of shares of Series B convertible preferred stock issued to our related parties in this financing:
|
Participants |
|
Shares of Series B preferred stock |
|
|
Aggregate purchase price ($) |
|
||
|
Entities affiliated with Perceptive(1) |
|
|
4,102,247 |
|
|
|
30,759,058 |
|
|
Entities affiliated with RTW(2) |
|
|
2,000,506 |
|
|
|
14,999,994 |
|
|
Osage University Partners III, LP(3) |
|
|
1,066,936 |
|
|
|
7,999,992 |
|
|
(1) |
Affiliates of Perceptive whose securities are aggregated for purposes of reporting share ownership information are Perceptive Life Sciences Master Fund, Ltd., Perceptive Xontogeny Venture Fund, LP and PX Venture (A), LLC. Perceptive is a beneficial owner of greater than 5% of our capital stock and is affiliated with our director Konstantin Poukalov. |
|
(2) |
Affiliates of RTW whose securities are aggregated for purposes of reporting share ownership information are RTW Master Fund, Ltd., RTW Innovation Master Fund, Ltd. and RTW Venture Fund Limited. RTW is a beneficial owner of greater than 5% of our capital stock and is affiliated with our director Dr. Roderick Wong. |
|
(3) |
Osage University Partners III, LP was a beneficial owner of greater than 5% of our capital stock at the time of this transaction. |
Participation in Initial Public Offering
In our initial public offering, certain of our directors, executive officers and 5% stockholders and their affiliates purchased an aggregate of 2,125,185 shares of our common stock. Each of those purchases was made through the underwriters at the initial public offering price. The following table sets forth the aggregate number of shares of our common stock that these 5% stockholders and their affiliates purchased in our initial public offering:
|
Participants |
Shares of common stock |
|
Aggregate purchase price ($) |
|
||
|
Entities affiliated with Perceptive(1) |
|
1,500,185 |
|
|
24,002,960 |
|
|
Entities affiliated with RTW(2) |
|
625,000 |
|
|
10,000,000 |
|
|
(1) |
Affiliates of Perceptive whose securities are aggregated for purposes of reporting share ownership information are Perceptive Life Sciences Master Fund, Ltd., Perceptive Xontogeny Venture Fund, LP and PX Venture (A), LLC. Perceptive is a beneficial owner of greater than 5% of our capital stock and is affiliated with our director Konstantin Poukalov. |
|
(2) |
Affiliates of RTW whose securities are aggregated for purposes of reporting share ownership information are RTW Master Fund, Ltd., RTW Innovation Master Fund, Ltd. and RTW Venture Fund Limited. RTW is a beneficial owner of greater than 5% of our capital stock and is affiliated with our director Dr. Roderick Wong. |
Employment Arrangements
We have entered into employment agreements or offer letter agreements with certain of our executive officers. For more information regarding our employment agreements with our named executive officers, see “Executive Compensation—Employment Agreements.”
Indemnification Agreements
We provide indemnification for our directors and executive officers so that they will be free from undue concern about personal liability in connection with their service to our company. Under our amended and restated bylaws, we are required to indemnify our directors and executive officers to the extent not prohibited under Delaware law. We have also entered into indemnity agreements with our executive officers and directors. These agreements provide, among other things, that we will indemnify the officer or director, under the circumstances and to the extent provided for in the agreement, for expenses, damages, judgments, fines and settlements he or she may be required to pay in actions or proceedings which he or she is or may be made a party by reason of his or her position as a director, officer or other agent of our company, and otherwise to the fullest extent permitted under Delaware law and our amended and restated bylaws.
138
Director Independence
Applicable Nasdaq rules, or the Nasdaq Listing Rules, require a majority of a listed company’s board of directors to be comprised of independent directors. In addition, the Nasdaq Listing Rules require that, subject to specified exceptions, each member of a listed company’s audit, compensation and nominating and corporate governance committees be independent and that audit committee members also satisfy independence criteria set forth in Rule 10A-3 under the Exchange Act of 1934, as amended, or the Exchange Act. The Nasdaq independence definition includes a series of objective tests, such as that the director is not, and has not been for at least three years, one of our employees, that neither the director nor any of his family members has engaged in various types of business dealings with us and that the director is not associated with the holders of more than 5% of our common stock. In addition, under applicable Nasdaq rules, a director will only qualify as an “independent director” if, in the opinion of the listed company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
Our board of directors has determined that all of our directors other than Dr. Bassaganya-Riera, representing four of our five directors, are “independent directors” as defined under applicable Nasdaq rules. In making such determination, our board of directors considered the current and prior relationships that each such director has with our company and all other facts and circumstances that our board of directors deemed relevant in determining his independence, including the beneficial ownership of our capital stock by each director and the transactions described in this section.
Item 14. Principal Accounting Fees and Services.
The following table represents aggregate fees billed to us for the fiscal years ended December 31, 2020 and 2019 by Ernst & Young LLP, our principal accountant.
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2020 |
|
|
2019 |
|
||
|
Audit Fees(1) |
|
$ |
950 |
|
|
$ |
95 |
|
|
Audit-Related Fees(2) |
|
|
|
|
|
|
|
|
|
Tax Fees(3) |
|
|
|
|
|
|
|
|
|
All Other Fees(4) |
|
|
|
|
|
|
|
|
|
Total Fees |
|
$ |
950 |
|
|
$ |
95 |
|
|
(1) |
Audit fees consist of fees billed for professional services provided in connection with the audit of our annual financial statements, the review of our quarterly financial statements and audit services that are normally provided by the independent registered public accounting firm in connection with regulatory filings. The audit fees also include fees for professional services provided in connection with our initial public offering in 2020. |
Pre-Approval Policies and Procedures
The Audit Committee has adopted a policy and procedures for the pre-approval of audit and non-audit services rendered by our independent registered public accounting firm, Ernst & Young LLP. The policy generally pre-approves specified services in the defined categories of audit services, audit-related services and tax services up to specified amounts. Pre-approval may also be given as part of the Audit Committee’s approval of the scope of the engagement of the independent auditor or on an individual, explicit, case-by-case basis before the independent auditor is engaged to provide each service. The pre-approval of services may be delegated to one or more of the Audit Committee’s members, but the decision must be reported to the full Audit Committee at its next scheduled meeting.
The Audit Committee has determined that the rendering of services other than audit services by Ernst & Young LLP is compatible with maintaining the principal accountant’s independence.
139
Item 15. Exhibits and Financial Statement Schedules.
Financial Statements
The following report and financial statements of the Company are included in this Annual Report on Form 10-K:
|
|
• |
Report of Independent Registered Public Accounting Firm |
|
|
• |
Consolidated Balance Sheets |
|
|
• |
Consolidated Statements of Operations and Comprehensive Loss |
|
|
• |
Consolidated Statements of Convertible Preferred Stock and Stockholders’ Deficit |
|
|
• |
Consolidated Statement of Cash Flows |
|
|
• |
Notes to Consolidated Financial Statements |
Financial Statements Schedules
All financial statement schedules have been omitted as they are not required, they are not applicable, or the required information is included in the financial statements or notes to the financial statements.
Exhibits
|
Exhibit Number |
|
Description |
|
3.1 |
|
|
|
3.2 |
|
|
|
4.2* |
|
|
|
10.1 |
|
|
|
10.2+ |
|
|
|
10.3+ |
|
|
|
10.4+ |
|
|
|
10.5+ |
|
|
|
10.6*+ |
|
|
|
31.1* |
|
|
|
32.1*## |
|
|
* |
Filed herewith. |
|
+ |
Indicates management contract or compensatory plan. |
|
## |
These certifications are being furnished solely to accompany this annual report pursuant to 18 U.S.C. Section 1350, and are not being filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, and are not to be incorporated by reference into any filing of the registrant, whether made before or after the date hereof, regardless of any general incorporation language in such filing. |
Not applicable.
140
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
LANDOS BIOPHARMA, INC. |
||
|
|
|
|
|
|
Date: March 31, 2021 |
By: |
|
/s/ Josep Bassaganya-Riera, Ph.D. |
|
|
|
|
Josep Bassaganya-Riera, Ph.D. |
|
|
|
|
Chairman, President and Chief Executive Officer |
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Josep Bassaganya-Riera, Ph.D. as his true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities, to sign this Annual Report on Form 10-K of Landos Biopharma, Inc., and any or all amendments thereto, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent full power and authority to do and perform each and every act and thing requisite or necessary to be done in and about the premises hereby ratifying and confirming all that said attorney-in-fact and agent, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signature |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/ Josep Bassaganya-Riera, Ph.D. |
|
Chairman, President and Chief Executive Officer |
|
March 31, 2021 |
|
Josep Bassaganya-Riera, Ph.D. |
|
(Principal Executive, Financial and Accounting |
|
|
|
|
|
Officer) |
|
|
|
|
|
|
|
|
|
s/ Jean-Frederic Colombel, M.D. |
|
Director |
|
March 31, 2021 |
|
Jean-Frederic Colombel, M.D. |
|
|
|
|
|
|
|
|
|
|
|
/s/ Christopher Garabedian |
|
Director |
|
March 31, 2021 |
|
Christopher Garabedian |
|
|
|
|
|
|
|
|
|
|
|
/s/ Konstantin Poukalov |
|
Director |
|
March 31, 2021 |
|
Konstantin Poukalov |
|
|
|
|
|
|
|
|
|
|
|
/s/ Roderick Wong, M.D. |
|
Director |
|
March 31, 2021 |
|
Roderick Wong, M.D. |
|
|
|
|
141
Exhibit 4.2
DESCRIPTION OF LANDOS BIOPHARMA, INC. COMMON STOCK
The following description of the common stock of Landos Biopharma, Inc., or the Company, and certain provisions of the Company’s amended and restated certificate of incorporation, or the restated certificate, and amended and restated bylaws, or restated bylaws, are summaries. These summaries are qualified in the entirety by reference to the provisions of the Delaware General Corporation Law and the complete text of the restated certificate and restated bylaws, which are incorporated by reference as Exhibits 3.1 and 3.2, respectively, of the Company’s Annual Report on Form 10-K to which this description is also an exhibit.
General
The restated certificate authorizes us to issue up to 200,000,000 shares of common stock, $0.01 par value per share, and 10,000,000 shares of preferred stock, $0.01 par value per share, all of which shares of preferred stock are undesignated. The Company’s board of directors may establish the rights and preferences of the preferred stock from time to time.
Common Stock
Voting Rights
Each holder of the Company’s common stock is entitled to one vote for each share on all matters submitted to a vote of the stockholders, including the election of directors. The affirmative vote of holders of at least 662/3% of the voting power of all of the then-outstanding shares of capital stock, voting as a single class, is required to amend certain provisions of the restated certificate, including provisions relating to amending the Company’s restated bylaws, the classified board, the size of the Company’s board, removal of directors, director liability, vacancies on the Company’s board, special meetings, stockholder notices, actions by written consent and exclusive forum.
Dividends
Subject to preferences that may be applicable to any then-outstanding preferred stock, holders of common stock are entitled to receive ratably those dividends, if any, as may be declared from time to time by the board of directors out of legally available funds.
Liquidation
In the event of the Company’s liquidation, dissolution or winding up, holders of common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of the Company’s debts and other liabilities and the satisfaction of any liquidation preference granted to the holders of any then-outstanding shares of preferred stock.
Rights and Preferences
Holders of common stock have no preemptive, conversion or subscription rights and there are no redemption or sinking fund provisions applicable to the common stock. The rights, preferences and privileges of the holders of common stock are subject to, and may be adversely affected by, the right of the holders of shares of any series of preferred stock that the Company’s board of directors may designate in the future.
Anti-Takeover Provisions
Section 203 of the Delaware General Corporation Law
The Company is subject to Section 203 of the Delaware General Corporation Law, which prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years after the date that such stockholder became an interested stockholder, with the following exceptions:
|
|
• |
before such date, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; |
|
|
• |
upon completion of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction began, excluding for purposes of determining the voting stock outstanding, but not |
|
|
the outstanding voting stock owned by the interested stockholder, those shares owned (i) by persons who are directors and also officers and (ii) employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
|
|
• |
on or after such date, the business combination is approved by the board of directors and authorized at an annual or special meeting of the stockholders, and not by written consent, by the affirmative vote of at least 662⁄3% of the outstanding voting stock that is not owned by the interested stockholder. |
In general, Section 203 defines a “business combination” to include the following:
|
|
• |
any merger or consolidation involving the corporation or any direct or indirect majority-owned subsidiary of the corporation and the interested stockholder; |
|
|
• |
any sale, transfer, pledge or other disposition of 10% or more of the assets of the corporation involving the interested stockholder (in one transaction or a series of transactions); |
|
|
• |
subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation or by any direct or indirect majority-owned subsidiary of the corporation of any stock of the corporation or of such subsidiary to the interested stockholder; |
|
|
• |
any transaction involving the corporation or any direct or indirect majority-owned subsidiary of the corporation that has the effect of increasing the proportionate share of the stock or any class or series of the corporation beneficially owned by the interested stockholder; or |
|
|
• |
the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits by or through the corporation. |
In general, Section 203 defines an “interested stockholder” as an entity or person who, together with the person’s affiliates and associates, beneficially owns, or within three years prior to the time of determination of interested stockholder status did own, 15% or more of the outstanding voting stock of the corporation.
Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws
The restated certificate provides for the Company’s board of directors to be divided into three classes with staggered three-year terms. Only one class of directors will be elected at each annual meeting of the Company’s stockholders, with the other classes continuing for the remainder of their respective three-year terms. Because the Company’s stockholders do not have cumulative voting rights, stockholders holding a majority of the shares of common stock outstanding will be able to elect all of the Company’s directors. The restated certificate and restated bylaws provide that directors may be removed by the stockholders only for cause upon the vote of 662/3% or more of the Company’s outstanding common stock. Furthermore, the authorized number of directors may be changed only by resolution of the board of directors, and vacancies and newly created directorships on the board of directors may, except as otherwise required by law or determined by the board, only be filled by a majority vote of the directors then serving on the board, even though less than a quorum.
The restated certificate and restated bylaws also provide that all stockholder actions must be effected at a duly called meeting of stockholders and will eliminate the right of stockholders to act by written consent without a meeting. The restated bylaws provide that only the Company’s Chairman of the board, Chief Executive Officer or the board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors may call a special meeting of stockholders.
The restated bylaws also provide that stockholders seeking to present proposals before a meeting of stockholders to nominate candidates for election as directors at a meeting of stockholders must provide timely advance notice in writing, and will specify requirements as to the form and content of a stockholder’s notice.
The restated certificate and restated bylaws provide that the stockholders cannot amend many of the provisions described above except by a vote of 662⁄3% or more of the Company’s outstanding common stock.
The restated certificates gives the Company’s board of directors the authority, without further action by the Company’s stockholders, to issue up to 10,000,000 shares of preferred stock in one or more series, with any rights, preferences and privileges as they may designate, including the right to approve an acquisition or other change in control.
The combination of these provisions will make it more difficult for the Company’s existing stockholders to replace the Company’s board of directors as well as for another party to obtain control of the Company by
replacing the Company’s board of directors. Since the Company’s board of directors has the power to retain and discharge the Company’s officers, these provisions could also make it more difficult for existing stockholders or another party to effect a change in management. In addition, the authorization of undesignated preferred stock makes it possible for the Company’s board of directors to issue preferred stock with voting or other rights or preferences that could impede the success of any attempt to change control of the Company.
These provisions are intended to enhance the likelihood of continued stability in the composition of the Company’s board of directors and its policies and to discourage coercive takeover practices and inadequate takeover bids. These provisions are also designed to reduce the Company’s vulnerability to hostile takeovers and to discourage certain tactics that may be used in proxy fights. However, such provisions could have the effect of discouraging others from making tender offers for the Company’s shares and may have the effect of delaying changes in the Company’s control or management. As a consequence, these provisions may also inhibit fluctuations in the market price of the Company’s stock that could result from actual or rumored takeover attempts. The Company believes that the benefits of these provisions, including increased protection of the Company’s potential ability to negotiate with the proponent of an unfriendly or unsolicited proposal to acquire or restructure the Company’s company, outweigh the disadvantages of discouraging takeover proposals, because negotiation of takeover proposals could result in an improvement of their terms.
Transfer Agent and Registrar
The transfer agent and registrar for the Company’s common stock is Broadridge Corporate Issuer Solutions, Inc. The transfer agent’s address is 1717 Arch St., Suite 1300, Philadelphia, Pennsylvania 19103.
Listing
The common stock is listed on the Nasdaq Global Select Market under the trading symbol “LABP.”
LANDOS BIOPHARMA, INC.
NON-EMPLOYEE DIRECTOR COMPENSATION POLICY
APPROVED BY THE BOARD OF DIRECTORS ON
March 30, 2021
Each member of the Board of Directors (the “Board”) of Landos Biopharma, Inc. (the “Company”) who is a non-employee director of the Company (each such member, a “Non-Employee Director”) will receive the compensation described in this Non-Employee Director Compensation Policy (the “Director Compensation Policy”) for his or her Board service.
The Director Compensation Policy may be amended at any time in the sole discretion of the Board or the Compensation Committee of the Board.
Annual Cash Compensation
Commencing at the beginning of the first calendar quarter following the closing of the initial public offering (the “IPO”) of the Company’s common stock (the “Common Stock”), each Non-Employee Director will receive the cash compensation set forth below for service on the Board. The annual cash compensation amounts will be payable in equal quarterly installments, in arrears following the end of each quarter in which the service occurred, pro-rated for any partial months of service. All annual cash fees are vested upon payment.
1.Annual Board Service Retainer:
(a)All Eligible Directors: $45,000
(b)Chairperson of the Board: $30,000
2.Annual Committee Chair Service Retainer:
(a)Chairperson of the Audit Committee: $20,000
(b)Chairperson of the Compensation Committee: $15,000
(c)Chairperson of the Nominating and Corporate Governance Committee: $10,000
3.Annual Committee Member Service Retainer:
(a)Member of the Audit Committee: $10,000
(b)Member of the Compensation Committee: $7,500
(c)Member of the Nominating and Corporate Governance Committee: $5,000
Equity Compensation
Equity awards will be granted under the Company’s 2019 Equity Incentive Plan, as amended (the “Plan”).
|
(a) |
Initial Appointment Equity Grant. On appointment to the Board, and without any further action of the Board or Compensation Committee of the Board, at the close of business on the day of such appointment, a Non-Employee Director will automatically receive a Nonstatutory Stock Option to purchase [36,000] shares of common stock. The Initial Grant shall vest in equal monthly installments such that the option is fully vested on the third anniversary of the grant date. |
|
(b) |
Automatic Equity Grants. Without any further action of the Board or Compensation Committee of the Board, at the close of business on the date of each Annual Meeting of the Company’s |
1
|
Stockholders, each person who is then a Non-Employee Director will automatically receive a Nonstatutory Stock Option to purchase 18,000 shares of common stock. Each Annual Grant will vest upon the one-year anniversary measured from the date of grant. |
|
(c) |
Vesting. All vesting is subject to the Non-Employee Director’s “Continuous Service” (as defined in the Plan) on each applicable vesting date. |
|
(d) |
Calculation of Value of a Nonstatutory Stock Option. The value of a Nonstatutory Stock Option to be granted under this Director Compensation Policy will be determined based on the Fair Market Value per share on the grant date. |
|
(e) |
Remaining Terms. The remaining terms and conditions of each Nonstatutory Stock Option, including transferability, will be as set forth in the Company’s standard Option Agreement, in the form adopted from time to time by the Board or the Compensation Committee of the Board. |
Expenses
The Company will reimburse Non-Employee Director for ordinary, necessary and reasonable out-of-pocket travel expenses to cover in-person attendance at and participation in Board and committee meetings; provided, that the Non-Employee Director timely submit to the Company appropriate documentation substantiating such expenses in accordance with the Company’s travel and expense policy, as in effect from time to time.
2
Exhibit 31.1
CERTIFICATION PURSUANT TO
RULES 13a-14(a) AND 15d-14(a) UNDER THE SECURITIES EXCHANGE ACT OF 1934,
AS ADOPTED PURSUANT TO SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Josep Bassaganya-Riera, Ph.D., certify that:
|
1. |
I have reviewed this Annual Report on Form 10-K of Landos Biopharma, Inc.; |
|
2. |
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
|
3. |
Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
|
4. |
I am responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
|
|
(a) |
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
|
|
(b) |
Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
|
|
(c) |
Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant's most recent fiscal quarter (the registrant's fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant's internal control over financial reporting; and |
|
5. |
I have disclosed, based on my most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant's board of directors (or persons performing the equivalent functions): |
|
|
(a) |
All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant's ability to record, process, summarize and report financial information; and |
|
|
(b) |
Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
|
Date: March 31, 2021 |
|
By: |
/s/ Josep Bassaganya-Riera, Ph.D. |
|
|
|
|
Josep Bassaganya-Riera, Ph.D. |
|
|
|
|
Chairman, President and Chief Executive Officer (Principal Executive, Financial and Accounting Officer) |
Exhibit 32.1
CERTIFICATION PURSUANT TO
18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report of Landos Biopharma, Inc. (the “Company”) on Form 10-K for the period ending December 31, 2020 as filed with the Securities and Exchange Commission on the date hereof (the “Report”), I certify, pursuant to 18 U.S.C. § 1350, as adopted pursuant to § 906 of the Sarbanes-Oxley Act of 2002, that:
|
|
(1) |
The Report fully complies with the requirements of section 13(a) or 15(d) of the Securities Exchange Act of 1934; and |
|
|
(2) |
The information contained in the Report fairly presents, in all material respects, the financial condition and result of operations of the Company. |
|
Date: March 31, 2021 |
|
By: |
/s/ Josep Bassaganya-Riera, Ph.D. |
|
|
|
|
Josep Bassaganya-Riera, Ph.D. |
|
|
|
|
Chairman, President and Chief Executive Officer (Principal Executive, Financial and Accounting Officer) |